
Review Article
Ann Hematol Oncol. 2019; 6(7): 1258.
Hyperhaemolysis Syndrome in Sickle Cell Disease
Win N*
NHS Blood and Transplant, UK
*Corresponding author: Nay Win, NHS Blood and Transplant, 75 Cranmer Terrace, London SW17 ORB, UK
Received: April 12, 2019; Accepted: July 04, 2019;Published: July 11, 2019
Abstract
Hyperhaemolysis Syndrome (HHS) is a potentially life-threatening complication of blood transfusion. It is a distinct complex syndrome resulting destruction of both the transfused and patient’s own RBC [1]. Although less common compared to patients with Sickle Cell Disease (SCD), HHS has also been reported in thalassaemia patients and patients with other haematological disorders. Fatal cases have been reported as a result of additional blood transfusion in patients with SCD [2,3] and a patient with underlying Myelofibrosis [4]. Therefore, correct diagnosis and prompt treatment is important.
Keywords: Hyperhaemolysis syndrome; Sickle cell disease
Introduction
The term “syndrome” was coined by Petz et al, [1] reported as “the sickle cell haemolytic transfusion reaction syndrome in 1997. At that time of writing, they have concluded that the pathogenesis is multifactorial and more definite data is required. New evidences added and updated the findings in this review article. The diagnostic criteria of HHS consists of four unusual features of haemolysis described by Petz et al, [1].
Firstly: “it may manifest as acute or delayed Haemolytic Transfusion Reactions (HTR), serological studies may not provide an explanation for the HTR. In some patients no alloantibodies are demonstrable or patients may have alloantibodies for which antigen-negative RBCs are readily obtainable. Even RBCs that are phenotypically matched with multiple patient antigens may be haemolysed”. These findings contradicted the basic principle of immunohematology. Providing cross matched compatible RBC units is to achieve a desired Hb increment and to avoid HTR. Following this, HHS is classified into acute and delayed forms, based on analysing 28 cases from 7 different publications in a review article published in 2008 (15 classified as acute and 13 as delayed form) [5].
Acute PTHS: occurs less than 7 days’ post-transfusion. The Direct Antiglobulin Test (DAT) is negative. There are no new red cell alloantibodies detected in follow-up serological investigations.
Delayed PTHS: usually occurs more than 7 days’ post-transfusion, (DAT) is positive. New alloantibodies identified [5].
Secondly: Petz [1] reported “that the patient developed a more severe anemia after transfusion and suggested that not only the transfused cells were haemolysed, but destruction of patient’s own RBC may play a role resulting in significant decrease in Hb level. King et al, [6] also proposed that sequential quantitation of HbA% and HbS% in the patient’s blood sample assist to capture the trajectory of haemolysis. Destruction of transfused and patient’s RBC was substantiated by serial analysis of urine by High Performance Liquid Chromatography (HPLC) demonstrating both the HbS (patient’s) and HbA (transfused RBC) in the urine by Win et al, [7] in 2001.
Thirdly: Petz at al, [1] observed a marked reticulocytopenia (a significant decrease from the patient’s usual absolute reticulocyte level) and recovery manifested by reticulocytosis and gradual improvement in Hb level. This is an unusual finding as a reticulocytosis is common presenting features of haemolysis (compensatory mechanism).
Fourthly: Not like classical Delayed Haemolytic Transfusion Reaction (DHTR) additional transfusion may further exacerbate haemolysis [1] and may become life-threatening or even may cause death [2-4].
Fifthly: After a recovery period, similar symptoms may recur following subsequent transfusion in some patients [1].
These unusual findings highlight the complexities of HHS and should be recognised as “syndrome” as defined by” Petz et al [1].
When describing HHS: Majority used the term “hyperhaemolysis syndrome” [4,5,8-16], followed by “hyperhaemolysis” [2,17-22] and
“(DHTR)” [23-28] the term DHTR / hyperhaemolysis syndrome [29,30] have also been used. Classical DHTR, [3,26] is a wellestablished complication of blood transfusion and it occurs as a result of immune destruction of the transfused RBC by red cell antibodies. Any recipient may develop classical DHTR. In general, it occurs between 2 to 21 days’ post-transfusion. Usually the DAT is positive with identification of red cell allo-antibodies that are not detected in the pre-transfusion sample. This is due to an anamnestic immune response as a result of re-exposure to red cell antigen which has previously been sensitized by transfusion, pregnancy or transplant. Reticulocytosis is common reflecting compensatory bone marrow erythropoiesis and subsequent transfusion with an antigen-negative unit may correct the anemia.
The incidence of HHS is not known and not all the patients who received blood developed HHS. Mwesigwa et al, reported a prevalence of 5% in SCD patients and commented that there is a potential role of recipient genetics in the susceptibility of HHS [31]. There is a striking difference in the clinical course, management and outcome between classical DHTR and HHS therefore it is crucial to distinguish between them. Some have commented that “there is no consensus definition of DHTR” when describing HHS [32] and even used the term “alloantibody –negative DHTR” without providing definite evidence [32]. Some described HHS as a subtype of DHTR [17]. Using the term DHTR when describing HHS in the literature is somewhat loose and confused. In our institution when describing HHS, we strictly adhere the “syndrome criteria“laid down by Petz et al, [1].
Although the first case of HHS was reported in 1993 in a SCD patient [2] uptill recently the pathogenesis still appears to be a subject of debate, [18] but most have cited the following hypotheses: i) marrow suppression, [1] ii) bystander mechanisms [6,18], and iii) macrophage activation [5,7,8]. The presenting symptoms are perplex and some commented that “bystander haemolysis and suppression erythropoiesis” occurs by unknown mechanism [33] and some suggested that reticulocytopenia is due to “accelerated destruction of reticulocytes as a result of selected antibody targeting of reticulocytes” [29].
Here we present the evidences related to the above three proposals and discuss the therapeutic intervention based on each of the theories and clinical outcome.
Firstly: “Marrow suppression theory”
One of the presenting features of HHS is relative reticulocytopenia and Petz et al, [1] have initially suggested that the apparent increase of the rate of haemolysis of autologous RBC was due to transfusion “suppression of erythropoiesis”.
They described [4] Delayed and one acute form of HHS. Each patient received additional transfusion of 7 to 8 units (mean 13 units) and were discharged on days [2,24,29,36] and [52] after the admission. As these patients received multiple transfusions over a certain period, it is possible that suppression of erythropoiesis might play some part in contributing to the worsening anemia. Despite providing cross matched compatible RBCs, there is an ongoing haemolysis and Petz et al, [1] recommended to withhold transfusion and to prescribe corticosteroid, as two recovered gradually with steroid therapy. In view of the reticulocytopenia, Erythropoietin (EPO) has been prescribed in HHS either as a single supportive therapy [6] or as a supplement to IVIG /steroids [17,26,29].
Response to steroids therapy contradicted the marrow suppression theory and it clearly indicates that another element may be involved in the pathogenesis of HHS. Therefore, in 2001 Win et al, [7] have proposed the alternative theory; that the “reticulocytes are destroyed by activated macrophage” (i.e. peripheral consumption). See subheading Macrophage activation theory.
Secondly: “Bystander mechanisms” [6,18]
Petz described bystander mechanism as ”Immune haemolysis of cells that are negative for the antigen against which the relevant antibody is directed” [34]. Therefore, in the process of antigen/ antibody reactions, implicated antibodies form immune complexes, complement activation, resulting reactive lysis for those RBCs lacking cognate antigen. Sickle RBCs show increased susceptibility to reactive lysis due to suggested functional defect of CD59. King et al, [6] reported 5 cases of HHS after receiving exchange transfusion of which 2 acute and 3 delayed forms. They conducted serial measurement of HbA and HbS levels in the peripheral blood and as a marker to document both the presence and absence of autologous destruction. Based on the evidence of autologous red cell destruction they have concluded “we have shown one patient with clinical evidence of bystander haemolysis complicating a DHTR”.6 No definite proof of laboratory evidence has been provided.
Bystander mechanism fails to cover or explain the following complex presenting symptoms of HHS:
i) As there was no antibody detected in acute HHS it is difficult to explain how the autologous RBCs were destroyed by bystander mechanism.
ii) In HHS despite providing antigen-matched crossmatchcompatible units (no potentiation for complement activation) haemolysis may occur.
iii) Petz et al, [1] have recommended”if possible to withhold further transfusion and to try oral prednisolone 1-2 mg/kg/day and to monitor closely.” It is obvious that steroid therapy will not resolve immune complex bystander mechanism of red cell destruction.
iv) Bystander theory fails to explain the reticulocytes findings in HHS; relative reticulocytopenia at presentation and a rise in reticulocyte count with recovery.
v) HHS has also been reported in Non SCD patients, who are not susceptible to reactive lysis.
“Anti-complement agent Eculizumab”. Eculizumab is a monoclonal antibody which inhibits complement activation by targeting C5, preventing progress into C5b-9 membrane complex. Sickle RBCs show increased susceptibility to reactive lysis due to suggested functional defect of CD59. Based on that, assumption has been made that bystander haemolysis might play a role in destruction of RBCs in HHS and Eculizumab has been tried. Gupta et al, [15] have tried Eculizumab in HHS and reported unresponsive to therapy. Dumas et al, [25] have tried Eculizumab as salvage therapy in patients with SCD. One received plasma exchange and another had liver transplantation. Delayed recovery was recorded in all cases and the authors have concluded that further assessments are required in prospective studies, taking into account the cost and possible side effects of this therapy.
Thirdly: ”Macrophage activation theory”
In acute HHS there is no evidence of red cell antibody mediated haemolysis and the activated macrophage theory focuses on the role of adhesion molecules, was proposed by Win et al. [5,7,8] depicted in (Figure 1) and (Figure 2) as follows:
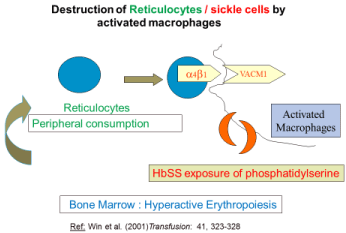
Figure 1: Explains how the patient’s own RBC (HbSS) and sickle reticulocytes
are destroyed via activated macrophage.
Destruction of patient’s own HbSS and reticulocytes in SCD [7,8] (Figure 1)
Hb SS adhere to macrophage more readily than HbA RBC through amino phosphatides (phosphatidylserine) express on the outer membrane of sickled RBC. Reticulocytes were also destroyed by adhesion mechanism. (Activated macrophages express Vascular Cell Adhesion Molecule [VCAM-1] which interacts with a4β1 integrin. a4β1 integrin is expressed in reticulocytes in SCD).
Peripheral consumption of reticulocytes was further supported by bone marrow studies and peripheral blood film findings. The bone marrow aspirate, examination conducted on a SCD patient presented with reticulocytopenia /HHS showed erythroid hyperplasia [7]. Danaee et al, [20] also reported erythroid hyperplasia in a bone marrow study of a patient with underlying HbH. That patient also expressed low reticulocyte count at the time of admission.
A peripheral blood film of a SCD patient admitted with HHS showed extremely poikilocytosis, abundant nucleated red cells and erythrophagocytosis by mononuclear cell [35].
Destruction of HbAA (transfused RBC) in SCD and in destruction of HbA/reticulocytes in Non SCD patients [5,8] (Figure 2)
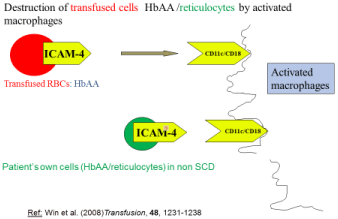
Figure 2: Explains a) how transfused RBC (HbAA) are destroyed via
activated macrophage in SCD patients b) how HbAA /reticulocytes destroyed
in Non SCD patients.
Ihanus et al, [36] (2007) have proposed that Intercellular Adhesion Molecule-4 (ICAM-4), a glycoprotein expressed on red blood cells and erythroid precursor cells, interacts with macrophages via integrin receptors CD11c/CD18.
HHS has also been reported in Non SCD patients and the presenting symptoms are the same, with destruction of both the transfused and autologous RBC with reticulocytopenia at the time of presentation. Based on the Ihanus [36] proposal.
Win et al, have suggested that the transfused HbAA in SCD and RBC/reticulocytes in Non SCD are destroyed by macrophages via adhesion mechanism [7,8] as depicted in (Figure 2).
HHS is not exclusive to SCD patients but also reported in non- SCD patients with the following disorders: myelofibrosis, [4] anemia of chronic disease, [14] mantle cell lymphoma, [12] dyserythropoietic anaemia, [37] CLL19 and other haemoglobinopathies such as: HbH disease, [20] β-thalassaemia major, [21,22,30] thalassaemia intermedia [10,11,38]. The majority of these patients have splenomegaly where macrophages are abundant.
What causes macrophage activation?
HHS occurs only in a subset of patients after transfusion of compatible blood. Mwesihwas et al, [31] have conducted the wholeexome sequencing of SCD patients with HHS and observed a rare, heterozygous stop-gain variant
(p. Glu210Ter) in MBL was significantly enriched among HHS cases. They have concluded that this group may be vulnerable to HHS.
We speculate [39] that transfusion with stored blood may trigger macrophage in these susceptible individuals.
Macrophage activation is negatively regulated via the CD47-sirpa which mediates a “don’t eat me” signal, but a change in conformation switches macrophages to an “eat me” mode and this change is seen in both stored blood and in sickle RBC [40,41]. Moreover HHS takes place in a heightened inflammatory state with increased level of inflammatory cytokines including TNF, IL1 and IL6, that rises further during vaso-occlusive crisis. Both IL1 and IL6 are activators of Macrophages [42].
We presented the following four additional evidences which support Macrophage activation [See as below i), ii), iii) and iv]
i) Serial measurement of ferritin
As Ferritin is released buy macrophages after erythrophagocytosis, we measured Ferritin as a non-specific biomarker for Macrophage activation. Ferritin study revealed a significant rise from baseline during haemolysis and decrease with recovery [35]. Serum Ferritin levels were 45079 μg/L,18927 μg/L,4772 μg/L, 1777 μg/L on days 1,6,9 and 18 of admissions. Similar findings were also reported in a CLL patient presented with HHS [19]. Serial measurement of serum Ferritin level may assist in reaching the diagnosis and may be a useful indicator of disease activity as described in two cases, reported in 2018 and 2019 respectively [43,44].
ii) Histopathological findings
In 2019 Win et al, have reported the histopathological findings of two cases [39] presented with acute form of HHS.
The first was a patient with underlying SCD who died of acute haemolysis within 48 hours of admission. The second was a pregnant patient with thalassaemia intermedia who developed resistant HHS (not responding to steroids/IVIG/Rituximab) which only resolved following laparoscopic splenectomy. Post-mortem findings for case #1 showed widespread erythrophagocytosis of red cells by activated macrophages with increase in numbers in bone marrow, liver and spleen. Erythroid hyperplasia was also reported in the bone marrow. For case #2 similar findings were demonstrated in the spleen tissue. Immunohistochemistry: Specific immunohistochemistry stain for monocyte and macrophage series CD68/PGM-1 was also prepared.
This was the first kind of histopathological findings reported in the literature which further supported the macrophage activation theory.
iii) Hb and reticulocyte response to anti-Macrophage therapy (IVIG/steroids)
The very first case of HHS was treated with IVIG/steroids from our institution in 1995.45 Several possible mechanisms on suppression on macrophage activity has been proposed by Win et al, [8,46]. Fever is one of the presenting features of HHS. IVIG is a pooled products and contains a broad range of antibacterial/antiviral specificity, which may neutralise infections and may thus reduce the activation of macrophages. IVIG might suppress hyperactive macrophages through immunomodulatory mechanism and IVIG may prevent adhesion interactions between sickle erythrocytes, reticulocytes and macrophages [46]. Steroids itself suppress macrophage activity [47]. Steroids and
IVIG may have a synergistic effect in suppressing macrophages [46].
Use of IVIG for HHS has been recommended by IVIG Hematology and Neurology Expert Panel (Canada) in 200748 followed by clinical guidelines for IVIG use (UK) in 201149 and guidelines on red cell transfusion in SCD in UK (2017), (British Committee for standards in Hematology) Davis et al, [50]. Based on these guidelines a combined IVIG /steroids therapy has been established as the first line therapy for HHS in UK. From our institution, we have previously reported nine cases [5,7,35,45,46,51,52] (six acute and three delayed form) of HHS in seven different publications. Information regarding IVIG/steroids therapy, days of treatment, transfusion support, reticulocyte count and Hb response to therapy were expressed in detail, as a Figure, for each published case. In seven cases, response was achieved within 4/5 days of initiating IVIG/steroids therapy. An increase in reticulocyte count together with a rise in Hb level were recorded in all of these cases. Seven patients required additional transfusion with IVIG/ steroids cover and in two patients (delayed form) transfusion was avoided [46,51] due to rapid response to IVIG/steroids treatment. One case (paediatric patient) presented with recurrent PTHS5 and additional course of IVIG/steroids was given and response achieved after day four of therapy and Hb level rose to 7.1g/dl.
Epo was only prescribed in two patients with definite indications. One with renal failure receiving peritoneal dialysis7 and another patient was prescribed due to human parvovirus B19 infection presented with transient red cell aplasia.52 Erythropoietin is a growth factor for both endothelial cells and erythroid cells. Trial et al, have reported that, “decreased levels of erythropoietin may trigger a process called neocytolysis”. Neocytolysis is the destruction of young red cells at the endothelial-macrophage interface and the authors have speculated that this is due to “decreased production of macrophage deactivating TGF-β by endothelial cells, resulting in activation of adhesions on macrophages which triggers phagocytosis of young red cells” [53]. We have previously proposed that there are two different mechanisms whereby EPO correct anemia in HHS [52]. Either by stimulating erythroid precursors or suppressing neocytolysis by macrophages. Although haemolytic process was halted after 5 days of IVIG/steroids therapy the patient needed further transfusion support as a result of parvo induced red cell aplasia.
Haemolysis, reticulocytopenia and peripheral consumption has been well described in patients with severe autoimmune haemolytic and EPO was not prescribed in these cases. Citation and explanation related to peripheral consumption in AIHA and reticulocytopenia has been fully discussed in one of Win et al, papers [5].
Response to a combination of IVIG/steroids in 7 cases (within 4 days of therapy), without EPO supplement, confirms there is no major element of marrow suppression. Based on these findings and evidence from AIHA, we concluded that there is no additional benefit achieved by prescribing EPO in HHS.
Serum erythropoietin levels are abnormally low for the extent of anemia in SCD [54]. Base line Erythropoietin should be checked before starting this treatment and EPO should be only prescribed if level is low or only with definite indications.
Infusion of IVIg has been associated with thrombosis and estimated 0.6% risk of stroke in non SCD patients [55]. We have taken a cautious approach and we provided a low dose IVIG regime (o.4 g/kg/day x 5 days) [5,7,35,45,46,51,52] (a total dose of 2g/kg). In a recent review article, Danee et al, [17] have reported 8 cases of PTHS with SCD from their institution (6 acute form and 2 delayed HHS). They have prescribed a high dose (1g/kg/day for 2 days), (a total dose of 2g/kg). There were no adverse events and have concluded that ”the combination of IVIG and steroids has always been successful in halting haemolysis thus far, only two patients require additional transfusion, and we have no need to use additional immunosuppressive medication”. Therefore, IVIG high dose regime appears safe and responses were achieved both in acute and delayed form of HHS. Optimal steroid dose is not well established. We prescribed IV methylprednisolone 0.5g/day to 1g/day (adults) and 4 mg/kg (paediatric patients) for 2 days and Dane et al, [17] prescribed Methylprednisolone 500mg daily for 3 days. Response to IVIG/steroids vindicated Petz’s observation of therapeutic value of prescribing steroids in HHS.
Not only that IVIG/steroids therapy is effective in HHS, it also prevents both the acute HTR and classical DHTR after transfusion with incompatible RBC units in non SCD patients [56,57].
It is possible that the some of the classical DHTR may progress into delayed form of HHS. In this scenario, transfused RBC were initially destroyed via antigen /antibody reactions through macrophage Fc receptor, but as a result of the activated macrophages, patient’s own RBCs and reticulocytes were also destroyed via adhesion mechanism,triggering delayed form of HHS, which resulted in a drop in Hb to below pre-transfusion level. Win et al reported a severe delayed from of HHS with multiple antibodies (anti-S,anti- Fy3 and anti-Jkb). The patient received exchange transfusion and immediate Post Exchange Hb level was 11g/L. After two days Hb dropped to 47g/L (presented with reticulocytepenia), and HHS was suspected and IVIG/steroids were commenced immediately, there was a rapid rise in reticulocyte count, and Hb response and the patient did not need additional transfusion [51]. Theoretically an assumption can be made that the patient’s own RBCs were destroyed via bystander mechanism, but this case clearly demonstrated that this was not the case. Response to IVIG/steroids support the macrophage activation theory as IVIG/steroids will neither block nor correct red cell destruction by bystander mechanism.
Since HHS has been well established as a separate syndrome, in UK, Serious
Hazard of Transfusion (SHOT, UK) an independent haemovigilance scheme has been collecting data on HHS [58]. Between 2010-2017, 30 cases reported, one patient had a recurrent HHS.
The first fatal case of HHS was reported in the 2010 SHOT report [3]. There were 6 cases reported in 2017, 5/6 symptoms improved with IVIG/methylprednisolone therapy but one patient died of acute chest syndrome. Although majority response to IVIG/steroids therapy patient may die of severe rapid acute haemolysis.
Recurrent HHS and preventive measures
Contrary to classical DHTR, in some patients HHS may recur despite providing antigen negative and crossmatch compatible units [1].
Petz et al, [1] presented 5 cases and 2/5 patients had recurrent PTHS. In the first case the patient had a recurrent PTHS twentyseven months later. Further transfusions were discontinued because of repeated episodes of PTHS. In the second case the patient had recurrent episode fifteen months after the first event. Transfusion was discontinued and steroids prescribed. Win et al, [5] reported a recurrent PTHS in a paediatric patient after a few weeks and Talano et al, [29] have reported a child who had 3 separate episodes of PTHS. Second and third episodes occurred after 6 and 12 months from the initial event respectively. The incidence of recurrence is not known, majority of SCD patients will require lifelong transfusion support and it is not possible to predict which patients may recur.
Macrophages are already activated in patients with SCD. But comparative studies among different patients showed both adherence to macrophages and erythrophagocytosis to be highly variable among SCD patients [59]. Even RBC survival studies in the same individual during two separate occasions at asymptomatic steady state demonstrates a variation in RBC destructions [60].
In our institution we prescribed IVIG prophylaxis to prevent recurrent PTHS since 2001 [61]. The same approach was also taken by Danee et al, [17].
Although HHS may recur, based on the variability of macrophage activation, it is not possible to predict when and which patient may recur after subsequent blood transfusion. Therefore, it is not possible to confirm or refute that prescribing IVIG successfully prevents the recurrence. Decision to prescribe IVIG as a precautionary measure is made on individual cases based on the severity of the previous episode of HHS.
In 2007, Noizat-Pirenne et al, [27] had tried Rituximab in patients with a history of HHS to prevent the recurrence. During the first episode the patient was treated with steroids, erythropoietin and cyclophosphamide. Two years later the patient required a hip replacement. On admission the patient was stable with no evidence of haemolysis. Rituximab was prescribed to cover transfusion for surgery and transfusion was uneventful. As like in IVIG therapy it is not possible to prove conclusively that Rituximab prevents recurrence of HHS.
Based on the above preventive case, attempt was made to treat HHS with Rituximab. In 2009, Bachmeyer [13] reported a case of HHS successfully transfused and concomitantly treated with a combination of Rituximab, steroids and EPO therapy in 2009. This was published as a correspondence and the title was given as “Rituximab as an effective treatment of HHS in SCD”. As the patient also received steroids and EPO it is difficult to claim that the effective therapy was as a result of Rituximab.
In 2013 Delmonte et al, [23] had reported a case titled ”IVIG resistance DHTR treated with rituximab in an adult sickle cell patient”. That case was unusual as the adult patient with SCD presented with PTHS associated with splenic sequestration. The patient did not respond to IVIG and single dose of Rituximab was prescribed on the day of splenectomy. Win [62] had commented that cessation of haemolysis immediately after splenectomy suggests that the response was likely to be due to removal of the site of red cell destruction (spleen). Therefore, it was difficult to prove that the desired response achieved within 24 hrs of prescribing Rituximab was a direct result of therapeutic outcome.
In 2015 Noizat-Pirenne et al, [28] had published an article “The use of Rituximab to prevent DHTR in SCD”. These are a group of seven SCD patients with a past history of severe DHTR (Hb dropped between 2 and 6 g/dl lower than immediate post-transfusion concentration). All had underlying alloantibodies except one. One patient gave a past history of four episodes of Hyperhaemolysis, another two episodes and the remainder, one episode. In this study two patients had further recurrent HHS despite providing extended antigen (Rh,K,Fy,jk and MNS) matched units. The study demonstrated that Rituximab may prevent additional antibody formation but it does not prevent recurrent haemolysis.
There is no conclusive evidence that Rituximab is an effective therapy for HHS. Rituximab causes depletion of B lymphocytes and there is a potential increase risk of infection and it has been suggested that the informed consent should be obtained from the patient before Rituximab is prescribed [62].
Resistant HHS
Majority will respond to prompt treatment with a combination of IVIG/steroids and some even might not need additional transfusion support as described by Danee et al, [17]. Having said that, some patients may not respond to initial dose of IVIG/steroids therapy and might need additional course of IVIG/steroids [5,7] or even may die of severe acute haemolysis. We define resistant HHS as those who do not respond to a second course of IVIG/steroids therapy.
Resistant HHS and spleen
In general, splenomagaly is unusual in adult patients with SCD due to autosplenectomy. Santos et al, [16] have reported three adult SCD patients presented with HHS responded to IVIG/steroids and EPO. Splenomegaly was reported in two patients, the one with a normal size and the other with a splenomegaly. Delmonte et al, [23] reported a resistant HHS presented with splenomegaly responded to splenectomy (i.e. removing the macrophage/site of destruction). Therefore, clinicians should be aware of the functional spleen when dealing with resistant cases. Splenectomy may only resolve HHS in some patients with thalassaemia [38,39].
Resistant HHS: plasma to RBC exchange
Uhlmann et al, [33] have reported a severe form of acute HHS not responding to IVIG/steroids, EPO and Rituximab therapy but was successfully managed with plasma to red blood cell exchange transfusion. They have concluded that it might be as a result of the removal of soluble mediators of haemolysis. It is possible that this process might remove “cytokines which might trigger the macrophage activation” [39].
Mortality and HHS
Patient may die of rapid haemolysis as a result of HHS. A patient with underlying Myelofibrosis [4] and a SCD39 died within 48 hrs of admission and receiving blood transfusion and both presented with acute form of HHS. Splenomegaly was recorded in both cases. The former was a 54 yr old patient who died of severe anemia Disseminated Intravascular Coagulation (DIC) and renal failure and the latter 26 yr old patient died of cardiac arrest, failed resuscitation. There was only one case report of death contributed by HHS in the SHOT report [3]. A pediatric patient with SCD was admitted with Hb of 5.4g/dl and 2 units were given.
Hb further dropped to 4.8g/dl and additional one unit was transfused. HHS was suspected, IVIG/steroids was prescribed and additional one unit transfused. Patient died the same day with multiorgan failure and Acute Respiratory Distress Syndrome (ARDS). There was one case reported each by Aygun et al, [63] and Elhusseini and Sabry [64] and the cause of death was reported as ARDS/DIC. The exact incidences are not known but appears to be rare. It is possible that some cases may not have been recognised as HHS and hence under-reporting.
Targeting Macrophage activation in HHS with Novel use of Toxilixumab
Patient with resistant HHS may die of severe rapid haemolysis despite treatment with IVIG/steroids and transfusion support as described in mortality section.
We have tried Tocilixumab (Macrophaghe specific therapy) in two SCD patients. In both cases, before prescribing Tocilixumab, Ferritin levels were checked. The former was reported by Lee et al, [43]. A 36-year-old SCD patient presented with a severe resistant HHS. Despite providing antigen negative and cross matched compatible units and with IVIG/steroids cover nadir Hb dropped to 2.1g/dl. DAT was negative and no new alloantibodies detected. At that point Ferritin was checked and there was a 100 fold rise in Ferritin level from base line 157ng/ml to 16,300ng/ml, which indicates macrophage activation. In view severity and not responding to IVIG/ steroids therapy, Toxilixumab, a humanized monoclonal antibody against the IL 6 receptor, which impaired macrophage function, was administered. As this is a novel therapy the consent was taken from the family. Ferritin immediately down trend, Hb stabilized and haemolysis resolved. The patient’s sample known to contain multiple antibodies (anti-e, anti-C, anti-K and anti-Fya) and for subsequent transfusion the patient was supported with hemoglobin-based oxygen carrier HBOC-201. The patient achieved complete recovery.
The latter [44] was a 33-year-old SCD patient with past history of two episodes of HHS. The patient was admitted for exchange transfusion and in view of the past history of HHS both IVIG/steroids were given to cover exchange transfusion. Despite that the patient had a recurrent (third episode) of HHS. High Ferritin, 18342 μg/L, was recorded and Toxilixumab prescribed. There was an increase in reticulocyte count and a decrease in LDH and Ferritin to 9923 μg/L.
There was a gradual rise in Hb level and a drop in Ferritin level. Measuring serum Ferritin level may assist in reaching the diagnosis and serial measurement also reflects as an indicator of disease activity. No adverse event noted in both cases with Toxilixumab therapy.
Conclusion
HHS is a complex syndrome. We adhere Petz et al, [1] diagnostic criteria when describing HHS. Clinicians should be aware that HHS and classical DHTR are two distinct disease entirety. More importantly in classical DHTR additional transfusion will correct anemia but in HHS it may further exacerbate haemolysis and even may cause death. High index of suspicion is important and any patients presented with haemolysis after transfusion with a drop in Hb below pre-transfusion should be regarded as a potential HHS. Serial measurement of reticulocytes, Ferritin and urine HPLC test will assist in reaching the diagnosis. If possible, avoid additional transfusion but, in severe rapid haemolysis, prompt treatment should be initiated with a combination of IVIG/steroids therapy and if indicated the patient should be transfused accordingly. We confirmed that the reticulocytes are destroyed by macrophages and there is no role for EPO in HHS. EPO should only be prescribed with definite indication or those presented with low level.
Management of patients with resistant HHS remains a problem. As discussed, Rituximab and Ecluzimab have been tried but need more firm evidences.
We presented the outcome of two patients with novel Macrophage specific therapy. Further study/clinical trials are needed to determine the optimal dose of Toxilixumab and to confirm the efficacy and safety of its use for treatment of life threating rapid acute haemolysis and resistant HHS cases.
References
- Petz LD, Calhoun L, Shulman I, Johnson C, Herron R. The sickle cell haemolytic transfusion reaction syndrome. Transfusion. 1997; 37: 382-392.
- Friedman DF, Kim HC, Manno CS. Hyperhaemolysis associated with red cell transfusion in sickle cell disease. Transfusion. 1993; 33: 148.
- Bolton-Maggs P. Transfusion Complications in Patients with Hemoglobin Disorders. Serious Hazards of Transfusion (SHOT) Annual Report. 2010.
- Treleaven JG, Win N. Hyperhaemolysis syndrome in a patient with Myelofibrosis. Hematology. 2004; 9: 147-149.
- Win N, New H, Lee E, De La Funete J. Hyperhemolysis syndrome in sickle cell disease: case report (recurrent episode) and literature review. Transfusion. 2008; 48: 1231-1238.
- King KE, Shirey RS, Lankiewicz MW, Young-Ramsaran J, Ness P. Delayed haemolytic transfusion reactions in sickle cell disease: simultaneous destruction of recipients’ red cells. Transfusion. 1997; 37: 376-381.
- Win N, Doughty H, Telfer P, Wild B, Pearson T. Hyper-haemolytic transfusion reaction in sickle cell disease. Transfusion. 2001; 41: 323-328.
- Win N. Hyperhemolysis syndrome in sickle cell disease. Expert Review of Hematology. 2009; 2: 111-115.
- Mechery J, Abidogun K, Crosfill F, Jip J. Hyperhaemolysis syndrome complicating pregnancy in homozygous of thalassaemia. Hemoglobin. 2012; 36: 183-185.
- Hebballi S. Post-Transfusion Hyperhaemolysis Syndrome (PTHS) in a pregnant patient with Beta Thalassaemia Intermedia. 2015; 1-379.
- Bezirgianninidoui Z, Christoforidou A, Kontekaki E, et al. Hyperhaemolytic Syndrome Complicating a Delayed Hemolytic Transfusion Reaction due to anti-P1 Alloimmunization, in a Pregnant Woman with HbO- Arab/ β-Thalassaemia. Mediterr J Hematol Infect Dis. 2016; 8: e2016053.
- Babb A, Diamantos N, Sekhar M. Hyperhaemolysis syndrome treated with corticosteriods and darbopoietin in a patient with mantle cell lymphoma. Transfusion Medicine. 2011.
- Bachmeyer C, Maury J, Parrot A, Bachir D, Stankovic K, Girot R, et al. Rituximab as an effective treatment of hyperhemolysis syndrome in sickle cell anemia. American Journal of Hematology. 2010; 85: 91-92.
- Darabi K, Dzik S. Hyperhemolysis syndrome in anemia of chronic disease. Transfusion. 2005; 45: 1930-1933.
- Gupta S, Fenves A, Taddie Nance S, Sykes DB, Dzik WS. Hyperhemoloysis syndrome in a patient without a hemoglobinopathy, unresponsive to treatment with eculizumab. Transfusion. 2015; 55: 623-628.
- Santos B, Portugal R, Nogueira C, Loureiro M. Hyperhemolysis syndrome in patients with sickle cell anemia: report of three cases. Transfusion. 2014.
- Danaee A, Inusa B, Howard J, Robinson S. Hyperhemolysis in patients with hemoblobinopathies: a single-center experience and review of the literature. Transfusion Medicine Review. 2015; 29: 220-230.
- Garratty G. What do we mean by “Hyperhaemolysis” and what is the cause? Transfusion Medicine. 2012; 22: 77-79.
- Rogers M, Smith G. Letter to Editor: Hyperhaemolysis in a patient with chronic lymphocytic leukemia. Transfusion Medicine. 2014; 24: 123-124.
- Danaee A, Howard J, Robertson B, Robinson S. Hyperhaemolysis in a patient with HbH disease. Transfusion Medicine. 2014; 4: 244-245.
- Grainger JD, Makar Y, McManus A, Wynn R. Refractory hyperhaemolysis in a patient with beta-thalassaemia major. Transfusion Medicine. 2002; 11: 55-57.
- Morawakage LR, Perera BJC, Dias PDN, Wijewardana SK. Hyperhemolysis in a patient with β- thalassemia major. Asian Journal of Transfusion Science. 2009; 3: 26-27.
- Delmonte L, Cantini M, Olivieri O, De Franceschi I. Immunoglobulin-resistant delayed hemolytic transfusion reaction treated with rituximab in an adult sickle cell patient. Transfusion. 2004; 53: 688-689.
- De Montalembert M, Dumont MD, Heilbronner C, Brousse V, Charrara O, Pellegrino B, et al. Delayed hemolytic transfusion reaction in children with sickle cell disease. Haematologica. 2011; 96: 801-807.
- Dumas G, Habibi A, Onimus T. Eculizumab salvage therapy for delayed hemolysis transfusion reaction in sickle cell disease patients. Blood. 2016; 127: 1062-1064.
- Gardner K, Hoppe C, Mijovic A, Thein SL. How we treat delayed haemolytic transfusion reactions in patients with sickle cell disease. British Journal of Haematology. 2015; 170: 745-756.
- Noizat-Pirenne F, Bachir D, Chadebech P, Michel M, Plonquet A, Lecron JC, et al. Rituximab for prevention of delayed hemolytic transfusion reaction in sickle cell disease. Haematologica. 2007; 92: 132-135.
- Noizat-Pirenne F, Habibi A, Mekontso-Dessap A, Razazi K, Chadebech P, Mahevas M, et al. The use of rituximab to prevent severe delayed haemolytic transfusion reaction in immunized patients with sickle cell disease. Vox Sanguinis. 2015; 108: 262-267.
- Talano JM, Hillery CA, Gottschall JL, Baylerian DM, Scott JP. Delayed haemolytic transfusion reaction/hyperhaemolysis syndrome in children with sickle cell disease. Pediatrics. 2003; 111: 661-665.
- Hannema SE, Brand A, van Meurs A, Smiers FJ. Delayed hemolytic transfusion reaction with hyperhemolysis after first red blood cell transfusion in child with beta-thalassemia: challenges in treatment. Transfusion. 2010; 50: 429-432.
- Mwesigwa S, Moulds JM, Chen A, Flanagan J, Sheehan VA, George A, et al. Whole-exome sequencing of sickle cell disease patients with hyperhemolysis syndrome suggests a role for rare variation in disease predisposition. Transfusion. 2018; 58: 726-735.
- Pirenne F, Yazdanbakhsh. How I safely transfuse patients with sickle-cell disease and manage delayed haemolytic transfusion reactions. Blood. 2018; 131: 2773-2781.
- Uhlmann EJ, Shenoy S, Goodnough LT. Successful treatment of recurrent hyperhemolysis syndrome with immunosuppression and plasma-to-red blood cell exchange transfusion. Transfusion. 2014; 54: 384-388.
- Petz LD, Garratty G. Immune hemolytic anemias. Philadelphia (PA). Churchill Livingstone. 2004; 159-160.
- Win N, Lee E, Needs M, Chia LW, Stasi R. Measurement of macrophage marker in hyperhaemolytic transfusion reaction: a case report. Transfusion Medicine. 2012; 22: 137-141.
- Ihanus E, Uotila LM, Tolvanen A. Red cell ICAM4 is a ligand for macrophage integrin CD11c/CD18. Blood. 2007; 109: 802-810.
- Bank I, Ermens AAM, Van Der Linden JM, Brand A. A life- threatening episode of treatment resistant haemolysis in a pregnant patient with dysertropoietic anaemia (CDA) type I. Transfusion Medicine. 2012; 22: 145-147.
- Vagace JM, Casado MS, Balo R, Gervasini G. Hyperhaemolysis syndrome responsive to splenectomy in a patient with thalassemia: a discussion on underlying mechanisms. Blood Transfusion. 2014; 12: 127-129.
- Win N, Lucas S, Hebbali S, McKernan A, Hamilton R, Robinson I, et al. Histopathological evidence for macrophage activation driving post-transfusion hyperhaemolysis syndrome. British Journal of Haematology. 2018.
- Burger P, Hilarius-Stokman P, de Korte D, van den Berg TK, van Bruggen R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood. 2012; 119: 5512-5521.
- Hillery CA, Scott JP, Du MC. The Carboxy-Terminal Cell-Binding Domain of Thrombospondin is Essential for Sickle Red Blood Cell Adhesion. Blood. 2000; 94: 302-309.
- Nnodim Johnkennedy, Meludu Samuel C, Dioka CE, Ifeanyichukwu M, UkaibeNkiru, Augustine I. Cytokine Expression in Homozygous Sickle Cell Anemia. Journal of Krishna Institute of Medical Sciences University. 2015; 4: 34-37.
- Lee LE, Beeler BW, Henderson AT, Graham BC, Osswald MB, Win N, et al. Targeting Macrophage Activation in Hyperhemolysis Syndrome with Novel Use of Tocilizumab. Blood. 2018.
- Linpower L, Silvapalaratnam S, Wisbey H, Jain S, Agapidou A, Win N, et al. Tociluzimab for the Management of Recurrent Resistant Post Transfusion Hyperhaemolysis in Sickle Cell Disease. Red Cell Disorders. 2019.
- Cullis JO, Win N, Dudley JM, Kaye T. Post transfusion hyperhaemolysis in a patient with sickle cell disease: use of steroids and intravenous immunoglobulin to prevent further red cell destruction. Vox Sanguinis. 1995; 4: 355-357.
- Win N, Tullie Y, Needs M, Chen FE, Okpala I. Use of intravenous immunoglobulin and intravenous methylprednisolone in hyperhaemolysis syndrome in sickle cell disease. Hematology. 2004; 9: 433-436.
- Packer JT, Greendyke RM, Swisher SN. The inhibition of erythrophagocytosis in vitro by corticosteroids. Transfusion Association of American Physicians. 1960; 73: 93-102.
- Anderson D, Kaiser A, Blanchette V, Brouwers M, Couban S, Radmoor P, et al. Guidelines on the Use of Intravenous Immune Globulin for Hematologic Conditions. Transfusion Medicine Reviews. 2007; 21: 9-56.
- Department of Health and Social Care. Clinical guidelines for immunoglobulin use: second edition update. 2011.
- Davis BA. Guidelines on red cell transfusion in sickle cell disease. Part 1: principles and laboratory aspects. British Journal of Hematology. 2007; 176: 179-191.
- Win N, Sinha S, Lee E, Mills W. Treatment with intravenous immunoglobulin and steroids may correct severe anemia in hyperhemolytic transfusion reactions: case report and literature review. Transfusion Medicine Reviews. 2010; 24: 64-67.
- Win N, Lee E, Needs M, Homeida S, Stasi R. Profound sustained reticulocytopenia and anemia in an adult patient with sickle cell disease. Transfusion Medicine. 2014; 24, 418-420.
- Trial J, Rice L. Erythropoietin withdrawal leads to the destruction of young red cells at the endothelial-macrophage interface. Current Pharmaceutical Design. 2004; 10: 183-190.
- Sherwood JB, Goldwasser E, Chilcote R. Sickle cell anemia patients have low erythropoietin levels for their degree of anemia. Blood. 1986; 67: 46-49.
- Caress JB, Cartwright MS, Donofrio PD, Peacock JE. The clinical features of 16 cases of stroke associated with administration of IVIg. Neurology. 2003; 60: 1822-1824.
- Win N, Needs M, Thornton N, Webster R, Chang C. Transfusions of least-incompatible blood with intravenous immunoglobulin plus steroids cover in two patients with rare antibody. Transfusion. 58; 1626-1630.
- Win N, Almusawy M, Fitzgerald L, Hannah G, Bullock T. Prevention of haemolytic transfusion reactions with intravenous immunoglobulin prophylaxis in U- patients with anti-U. Transfusion. 2019.
- Bolton-Maggs P. Haemolytic transfusion reactions. Serious Hazards of Transfusion (SHOT) Annual Report. 2017.
- Hebbel RP, Miller VB. Phagocytosis of sickle erythrocyte: immunological and oxidative determinants of haemolytic anemia. Blood. 2019; 64: 733-741.
- Monocyte P. Phagocyte activity in sickle cell disease. Acta Haematol. 1991; 85: 199-201.
- Win N, Clark M, Pollard C, Bevan D. Prevention of recurrence of hyperhaemolytic transfusion reaction in Sickle Cell Disease. Transfusion Medicine. 2001; 11: 13.
- Win N. Immunoglobulin-resistant delayed haemolytic transfusion reaction treated with rituximab in an adult sickle cell patient. Transfusion. 2013; 53: 2829-2830.
- Aygun B, Padmanabhan B, Paley C, Chandrasekaran V. Clinical significance of RBC alloantibodies and autoantibodies in sickle cell patients who received transfusions. Transfusion. 2002; 42: 37-43.
- El-Husseini A, Sabry A. Fatal hyperhemolytic delayed transfusion reaction in sickle cell disease: a case report and literature review. American Journal Emerg Med. 2010; 88: 1062-1068.