
Review Article
Ann Hematol Oncol. 2019; 6(8): 1262.
Change of PVSG-WHO Into The European Clinical Laboratory Molecular and Pathological (2019 CLMP) Criteria for Classification and Staging of JAK2, MPL and CALR Mutated Myeloproliferative Neoplasms: Bone Marrow Characteristics from Dameshek to Georgii, Thiele & Michiels
Michiels JJ1,2*, De Raeve H2, Popov VM2, Trevet M2 and Trifa A4
1International Hematology and Blood coagulation Research Center, Goodheart Institute and Foundation in Nature Medicine, Netherlands
2Department of Hematology, Colentina Clinical Hospital, Romania
3Department of Pathology, University Hospital Brussels and OLV Hospital Aalst, Belgium
4Department of Molecular Biology, ‘LiliuHatieganu’, University of Medicine and Pharmacy, Romania
*Corresponding author: Jan Jacques Michiels, Department of Hematology and Blood Coagulation Research Center, Goodheart Institute and Foundation in Nature Medicine, and International Collaboration and Research on Myeloproliferative Neoplasms: ICAR. MPN, Erasmus Tower Veenmos 13, 3069 AT Rotterdam, Netherlands
Received: June 03, 2019;Accepted: July 10, 2019; Published: July 17, 2019
Abstract
The one cause hypothesis of Dameshek for trilinear PV has been confirmed William Vainchenker in 2005 by his discovery of the acquired somatic JAK2V617F mutation as the cause of three clinical phenotypes of MPN ET, PV and MF. The Romanian Working Group on Myeloproliferative Neoplasms (RWG. MPN) changed the 1975 PVSG, 2008 WHO, and the 2015 European Clinical, Molecular and Pathological (ECMP) into the 2019 Clinical Laboratory, Molecular and Pathological (CLMP) classification. The RWG MPN defined in 2016 a broad spectrum of JAK2V617F mutated MPN phenotypes: normocellular ET, hypercelluar ET due to increased megakaryopoiesis is and erythropoiesis (EM in prodromal PV), hypercellular ET with Megakaryocytic-Granulocytic (EMG) trilinear myelo proliferation and various degrees of splenomegaly in erythrocythemic PV, early PV, classical PV, masked PV, advanced PV with MF and post-PV MF. ET heterozygous for the JAK2V617F mutational is associated with low JAK2 mutation load and normal life expectance. PV patients are hetero-homozygous versus homozygous for the JAK2V617F mutation in early vs advanced stages of PV with increasing JAK2 mutation load from below 50% to 100%, which is associated with increase of MPN disease burden during lifelong follow-up in terms of symptomatic splenomegaly, constitutional symptoms, bone marrow hyper cellularity and secondary MF. Pre-treatment bone marrow biopsy in prefibrotic MPNs are of diagnostic and prognostic importance because each of the JAK2, MPL and CALR MPNs are featured by a normocellular megakaryocytic stage followed by hypercellular stage with increasing grades of myelofinbosis. JAK2 exon 12 mutated MPN is a distinct benign early stage PV. CALR mutated hypercellular thrombocythemia show distinct PMGM bone marrow characteristics of clustered larged immature dysmorphic megakaryocytes with bulky (bulbous) hyper chromatic nuclei, which are not seen in JAK2 mutated ET and PV. MPL515 mutated normocellular thrombocythemia is featured by clustered giant megakaryocytes with hyperlobulated stag-horn-like nuclei without features of PV in blood and bone marrow. Myeloproliferative disease burden in each of the JAK2, CALR and MPL MPNs is best reflected by the degree of anemia, splenomegaly, mutation allele burden, bone marrow cellularity and myelofibrosis.
Keywords: Myeloproliferative neoplasms; Essential thrombocythemia; Polycythemia vera; Primary megakaryocytic granulocytic myeloproliferation; Myelofibrosis; JAK2V617F mutation; MPL515 mutation; Calreticulin mutation; JAK2 wild type; Bone marrow pathology
Introduction
The clinical characteristic, which should be present for a definite diagnosis of PV anno 1940 included plethoric appearance, splenomegaly, definitely elevated erythrocyte count above 6×1012/L, elevated platelet count, and elevated hematocrit [1,2]. The bone marrow is pathognomonic diagnostic showing large megakaryocytes and a panmyelosis of increased trilinear erythrocytic megakaryocytic granulocytic myeloproliferation [1,2]. Blood volume estimation (Red Cell Mass: RCM) was not required to diagnose PV in the studies of Dameshek [1-3]. Dameshek (1900-1969) (Figure 1) [2] considered the majority of PV patients as fundamentally normal and the treatment of PV should be venesection aiming at haematocrit of 0.40 resulting in a state of iron deficiency [1-3]. In PV in complete remission by phlebotomy alone red cell count remains elevated above 6×1012/L, but the haemoglobin and hematocrit levels remain low due to iron deficiency induced microcytosis of red cells for periods of months to years [1-4]. It is possible to relief symptoms and control hyper volume enemia in PV patients by phlebotomy alone for several to more than fifteen years. Such PV patient is in as good health as comparable persons of the same age group [3-5]. PV is a total marrow disorder of trilinear Erythrocythemic, Thrombocythemic and Granulocyte Micmyeloma Proliferation (EMGM) with blood erythrocytosis, leukocytosis and thrombocytosis [2]. Dameshek (1950) proposed the one cause hypothesis for PV as a trilinear Myeloma Proliferative Disease (MPD) due to either the presence of excessive bone marrow stimulation by an unknown factor or the lack or diminution of an inhibitory factor [2,3]. The one cause hypothesis of Dameshek for trilinear PV has been confirmed William Vainchenker (Figure 1) in 2005 by his discovery of the acquired somatic JAK2V617F mutation as the cause of Erythrocythemic, Megakaryocythemic and Granulocythemic Myeloproliferation (EMGM) associated with three clinical phenotypes of MPN Essential Thrombocythemia (ET), PV and myeloid neoplasia of the spleen with secondary Myelofibrosis (MF) [2,3]. Dameshek recognized in 1951 Megakaryocyte Leukemia (ML), which is consistent with Thrombocythemia associated with Primary Megakaryocytic Granulocytic Myeloproliferation (PMGM) as a distinct MPN entity recognized by Michiels in 2013 as CALR mutated thrombocythemia and myelofibrosis without features of PV (Figure 1) [6-9].
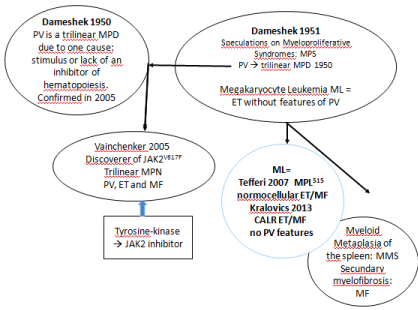
Figure 1: The 1950 Dameshek one cause hypothesis of trilinear PV and
Vainchenker’s discovery in 2005 of heterozygous and homozygous JAK2V617F
mutations as the driver cause of the trilinear erythrocytic, megakaryocytic
granulocytic myeloproliferation (EMGM) myeloproliferative neoplasms
polycythemia vera (PV, essential thrombocythemia (ET)and secondary
myelofibrosis (MF). In the editorial on speculations of myeloproliferative
esyndromes in 1951, Dameshek recognized megakaryocytic leukemia (ML)
as a distinct entity without features of PV at diagnosis and follow-up. This ML
entity has recently be recognized by Michiels as MPL515 or calreticulin (CALR)
mutated thrombocythemia without features of PV.
Change of Crude PVSG/WHO Into European Clinical, Molecular and Pathological (ECMP) Criteria for Et vs . PV
The 1975 PVSG and 2008 WHO criteria are crude with regard to normal level of Hemoglobin (Hb) level above 18.5 g/dl and Hematocrit (Ht) above 0.60 in men and Hb>16.5 and Ht>0.56 in women to diagnose PV with the need to measure Red Cell Mass (RCM). RCM measurement was the only criterion to distinguish JAK2V617F mutated ET from PV in cases with Hemoglobin (Hb) and Hematocrit (Ht) in the upper level of normal [10-12]. The 2013- 2015 ECMP criteria used bone marrow histology and mutation analysis as a pathognomic clue to each of the MPDs obviating the need to measure RCM to distinguish PV from primary or secondary erythrocytosis and.to distinguish ET from reactive thrombocytosis, from BCR/ABL positive thrombocythemia in CML and from thrombocythemia in Myelodysplastic Syndromes (MDS), 5q minus syndrome in particular by the demonstration of clustered mature large megakaryocytesin MPN and small megakaryocytes in CML and MDS [7-9]. Megakarycyte morphology are not different in prefibrotic JAK2V617 F mutated ET and PV patients (Figure 2, Tables 1 and 2).

Figure 2: Translation of the 2008 WHO clinical criteria into2018 CLMP criteria
for the classification of JAK2V617F mutated ET, PV and EMGM or masked
PV (red), versus CALR mutated primary mega karyocytic granulocytic
myeloproliferation (PMGM, blue) and MPL515 mutatednormocellular ET
(black). All molecular variants of MPN disease burden is reflected by the
degree of anemia, myeloid neoplasia of the spleen (splenomegaly), bone
marrow cellularity and secondary myelofibrosis.
The prospective Rotterdam studies assessed the ECMP criteria of PV (Table 2) related to Red Cell Mass (RCM), Hb, Ht and erythrocyte counts in 10 ET and 16 PV patients in whom RCM, peripheral blood and bone marrow data were available (Table 3). The correlation curves between erythrocyte count, Hb or Ht versus RCM showed the best correlation between erythrocyte counts and RCM (Figure 3). At RCM above 30 ml/kg the erythrocytes are above 5.8x1012/L in all 19 ECMP defined PV patients (Table 3, Figure 3). At erythrocyte counts above 5.8x1012/L the hematocrit values range from 0.46 to 0.72 in ECMP defined PV (Table 3, Figure 3). At erythrocyte counts below 5.8 x1012/L the hematocrit values range from 0.40 to 0.45 in ECMP defined ET who had normal RCM (Figure 3, Table 1). At erythrocytes above 5.8 x1012/L in PV patients the Hb values ranged from 15.0 to 20.9 and are below 2008 WHO criteria in 3 females and 2 males (Table 3 in blue), who had increased RCM. At erythrocytes above 5.8x1012/L in PV patients the Ht values ranged from 0.46 to 0.72 and are below 2008 WHO criteria but had increased RCM in 7 females and 1 male (Table 3 in blue). Seven ET patients had normal RCM at erythrocyte counts between 4.4 to 5.3 x1012/L of whom 4 had normocellular (‹60%) ET and 3 had hypercellular (60-80%) prodromal PV bone marrow histology (Table 3). Increase of erythrocytes counts above 5.8x1012/L for the diagnosis of PV appears to be independent from the iron deficient status and persists in PV in a clinical remission obtained by repeated venesection (Figure 4) thereby confirming the observations of Dameshek [2,3]. Erythrocyte count at a cutoff level of the upper limit of normal (5.8 x1012/L in males and 5.6 x1012/ L in females) separates ET and prodromal PV from classical PV (Figures 3 and 4, Tables 1 and 2) obviating the need to measure RCM in JAK2V617F and exon 12 mutated MPN patients. It is the degree of erythrocythosis (erythrocyte count above the upper limit of normal) on top of characteristic bone marrow histology, increased LAP score and decreased serum EPO levels that separates JAK2V617F mutated ET and prodromal PV from classical PV with the need of phlebotomy [7-9]. The reduction in iron reserve in PV leads to an insufficient amount of iron for the synthesis of haemoglobin in the developing red cells, and as a result that bone marrow iron stain is negative in PV [2,3], but usually present in ET [6-9]. As iron deficiency develops in PV on treatment with phlebotomy, the mature red cells produced become smaller (microcytic) than normal and occupy less room in the circulation, which is associated with the relief of hypervolemic symptoms. The hemoglobin and hematocrit levels remain low at Mean Red Cell Volumes (MCV) below 70fl for periods of months to years in PV patients in complete haematological remission by phlebotomy alone, but the erythrocyte count persist to remain above 5.8x1012/L (Figure 4). As the MCV of red cells becomes reduced to levels below 70 cubic micron due to the chronic iron deficiency state, the discrepancy between the high red cell count far above 6x1012/L and low hemoglobin level appears to be a diagnostic clue to PV in remission [2,3,6-9].
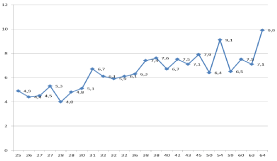
Figure 3: Erythrocyte counts x1012/L vertical axis versus red cellmass
(RCM) horizontal axis according to Michiels et al 1985-2018 (Tables 1
and 2). At erythrocyte values below 5.8 x1012/L the red cellmass (RCM)
values are between 25 and 30 ml/kg in essential thrombocythemia (ET). At
erythrocyte values above 5.7x1012/L all values of RCM area bove 30/kg in all
polycythemia vera (PV) cases indicating that the erythrocyte cut-off level of
5.7 x1012/L discriminates between ET and PV The numbers in the blue line
are erythrocyte counts x1012/L
![]()
Clinical laboratory molecular: CLM
Bone marrow pathology: P criteria
ET
Normocellular ET with M bone marrow
1. Platelet count of >350 x109/l and the presence of large platelets in a blood smear
2. Heterozygous JAK2-V617F mutation, and low JAK2 allele mutation load
3. Normal erythrocytes <5.8x1012/L males, <5.6 x1012/L females
4. Hemoglobin (Hb) and hematocrit (ht) normal or upper range of normalMegakaryocytic (M) myeloproliferation and clustering of enlarged mature pleomorphic megakaryocytes with hyperlobulated nuclei and mature cytoplasm, lacking conspicuous morphological abnormalities.
Normocellular bone marrow (<60%) and no proliferation or immaturity of granulopoiesis and no orsome increase of erythropoiesis.
Reticuline fibrosis (RF) 0 or 1prodromal PV
ET with EM bone marrow features of PV
Table 1: 2019 Clinical Laboratory Molecular and Pathobiological (CLMP) criteria for diagnosis of JAK2V617F mutated essential throbocythemia (ET) [6-9].
![]()
Clinical laboratory molecular: CLM
Bone marrow pathology: P criteria
Major criteria for PV
A 1. Hematocrit>0.51/>0.48 in male/female Erythrocytes >5.8x1012/L males >5.6x1012/L females
A 2. Presence of heterozygous and/or homozygous JAK2V617F or JAK2 exon 12 mutation
A 3. Low serum Epo level
Minor
B 1. Persistent increase of platelet count x109/L: grade I: 400-1500, grade II: >1500.
B 2. Granulocytes >10 x109/l or Leukocytes >12 x109/l and raised LAP-score or increased CD11b expression in the absence of fever or infection
B 3.Myeloid neoplasia of the spleen à splenomegaly on ultrasound echogram (>12 cm length in diameter) or on palpation.
B 4. Spontaneous endogenous erythroid colony (EEC) formation (optional)
P1. Bone marrow pathology: increased cellularity (60-100%) due to variable degrees but usually trilinear erythrocytic, megakaryocytic and granulocytic (EM or EMG) myeloproliferation (EMGM or panmyelosis according to Dameshek) and clustering of small to giant (pleomorph) megakaryocytes with hyperlobulated nuclei.
Absence of stainable iron. No pronounced inflammatory reactionP2. Erythrocytosis. Normal erythropoiesis, normal granulopoiesis and megakaryocytes of normal size, morphology and no clustering
Grading of secondary reticulin fibrosis (RF) and myelofibrosis (MF) [44-47].
Prefibrotic: RF-0/1 = MF-0Early fibrotic: RF-2 = MF-1
Fibrotic: RCF 3 = MF-2
Post-PV MF: RF4 = MF-3
2019 CLMP criteria for staging of prodromal, erythrocythemic, and advanced PV
A2 + B1 + P1 establish early PV (mimicking ET) prodromal PV CMP stage 0
A1 + A2 + A3 + P1 and none of B establish idiopathic erythrocythemia (IE) or stage 1 PV
A1 + A2 + A3 + P1 and one or more of B establish classic stages of PV stage 2 and 3
A2 + B3 + P1 detect masked cases of PV with splenomegaly and hypersplenism to be labelled as Inapparent PV (IPV) frequently seen Budd-Chiari syndrome or splanchnic vein thrombosis
Table 2: 2019 Clinical Laboratory Molecular and Pathological (CLMP) criteria for the diagnosis of prodromal, masked and classical JAK2 mutated polycythemia vera (PV) versus primary or secondary erythrocytoses [6-9].
![]()
ET PV
Age M/F
Hbmmol/L
Ht
Eryx109/L
RCM ml/kg
Hb g/dL
Pltx109/L
WBC x109/L
BM Iron
BM histology
1 ET
56 M
8.5
0.4
4.5
27
13.6
575
7
Pos
ET
2 ET
46 M
8.3
0.4
4.4
26
13.2
939
16
Pos
ET
3 ET
60 F
9.7
0.45
5.3
27
15.5
814
7
Pos
ET
4 ET
37 M
8.4
0.42
4
28
13.4
699
18
Pos
ET
5 ET
58 M
10
0.45
5.1
30
16
810
10
Neg
PV
6 ET
47 F
8.9
0.44
4.8
28
16.3
553
8
Neg
PV
7 ET
31 F
8.6
0.41
4.9
25
17.8
576
6
Neg
PV
8 ET àPV
60 F
10.4
0.49
6.7
31
16.6
792
10
Neg
PV
9 ETàPV
72 F
9.4
0.46
6.1
32
15
1436
13
Neg
PV
10 ETàPV
44 F
10.5
0.49
5.9
32
16.8
1304
14
Neg
PV
1 PV
43 F
10.8
0.52
6.1
32
17.2
405
14
Neg
PV
2 PV
50 M
11.6
0.63
6.3
36
18.5
397
7
Neg
PV
3 PV
47 F
10.2
0.53
7.4
38
16.3
924
13
Neg
PV
4 PV
38 M
11.1
0.6
6.7
40
17.8
384
8
Neg
PV
5 PV
63 M
11.1
0.56
6.5
59
17.8
1932
10
Neg
PV
6 PV
60 F
13.4
0.68
7.9
45
21.4
1065
17
Neg
PV
7 PV
49 F
10.9
0.57
7.5
60
17.4
728
8
Neg
PV
8 PV
66 M
12.2
0.64
7.1
63
19.5
1035
14
Neg
PV
9 PV
71 M
13.3
0.7
6.4
50
21.2
1320
16
Neg
PV
10 PV
65 M
11.9
0.65
7.6
38
19
1300
18
Neg
PV
11 PV
55 F
12.1
0.61
7.1
43
19.3
1085
13
Neg
PV
12 PV
59 F
11
0.59
7.5
42
17.6
708
17
Neg
PV
13 PV
74 F
13.1
0.72
9.1
54
20.9
959
9
Neg
PV
14 PV
71 M
12.5
0.66
9.9
64
20
609
18
Neg
PV
15 PV
66 F
9.5
0.51
6.7
33
15.2
646
18
Neg
PV
16 PV
44 F
10.5
0.49
5.9
32
16.8
1302
14.5
Neg
PV
Table 3: The relation between RCM, erythrocyte count and bone marrow histology findings at time of diagnosis in in 26 MPN patients: 10 ET and 14 PV and in 2 ET cases at time of evolution into PV as compared to the 2008 WHO cut-of levels of hemoglobin (Hb) and hematocrit (Ht) for PV: Hb >18.5 g/dl and Ht>0.60 in men and Hb>16.5 and Ht>0.56 in women for the diagnosis of PV. Michiels Personal Observations 1975-1985.
JAK2V617F Mutated Trilinear PV and ET: Dameshek-Vainchenker’s Disease
The one cause hypothesis of Dameshek for trilinear PV has been confirmed William Vainchenker (Figure 1) in 2005 by his discovery of the acquired somatic JAK2V617F mutation as the cause of three clinical phenotypes of MPN ET, PV and MF. The JAK2V617F mutated trilinear MPN is featured by Erythrocytic, Megakaryocytic and Granulocytic Myeloprolifeation (EMGM) with a broad spectrum of variable clinical manifestations including normocellular ET, prodromal PV, erythrocythemic PV with normal platelet and leukocyte count, classical PV, masked PV, and various degrees of myeloid neoplasia of the spleen and secondary MF [13,14]. The morphology of clustered medium to large megakaryocytes in bone marrow smears and biopsies were not different in JAK2V67F mutated ET and PV patients (Figure 5, Tables 1 and 2). Detection of JAK2V617F has become the first intention diagnostic test to differentiate between PV and Idiopathic Erythrocythemia (IE) from erythrocytosis with a sensitity of 95% and specificity of 100% [15-26]. The prevalence of the JAK2V617F mutation in PVSG defined PV is 95% and about 50% in ET and MF7 [15,16]. The majority of ET patients are heterozygous for the JAK2V617F mutation with a JAK2V617F mutation load of less than 10% to 50% of the granulocytes. Early stage PV patients are heterohomozygous for the JAK2V617F mutation with a mutation load of less than 50%, whereas PV patients with advanced MPN disease burden are homozygous for the JAK2 mutation with increased JAK2 burden between 50% to 100% of the granulocytes (Figure 6) [17-21]. A group of JAK2V617F positive normocellular ET with a very low percentage of heterozygous mutant JAK2V617F can maintain as a non-progessive subpopulation in the bone marrow without a tendency to evolve into prodromal PV or hypercellular ET during long term follow-up [22]. Patients with hypercellular ET and PV homozygous for the JAK2V617F mutation patients are at high risk for anemia and myeloid metaplasia of the spleen (splenomegaly) with secondary myelofibrosis [19]. Two studies demonstrated that so-called heterozygous PV with allele load less than 50% are hetero/homozygous at the EEC level in blood and bone marrow for the JAK2V617F mutation, whereas ET patients are heterozygous reflecting a maximal JAK2V617F mutation load of 50% [20,21]. Homozygosity for JAK2V617F results from mitotic recombination and homozygous-mutant BFU-E were present in most patients with PV but not in those with ET. According to Michiels & Vainchenker in 2006 heterozygous JAK2V617F mutation is enough to constitutively activate megakaryocytes due to with increased sensitivity to TPO for the induction of the clinical ET phenotype. The JAK2V617F mutated platelets are constitutively activated, hypersensitive and sticky platelets as the cause of plateletmediated plateletmediated aspirin-responsive Sticky Platelet Syndrome (Table 4) [23-25]. According to the Vaincheker’s“dosage” hypothesis the level and duration of JAK2V617F directly contribute to the phenotypic diversity of JAK2V617F mutated EMGM manifestations (Table 4). This hypothesis is based on different densities of TPO Receptors (TPOR or MPL) and EPO Receptors (EPOR) on hematopoietic progenitor cells and on differences of response of TPOR and EPOR to various levels of JAK2V617F activity [26,27]. MPL is expressed at high levels in megakaryocytic cells where it controls physiological TPO levels. It is possible that activation of TPO receptors by low levels of heterozygous JAK2V617F is sufficient to send a signal to megakaryocytic cells [13,16,28] (Table 4). Conversely, EPOR is expressed at low levels on hematopoietic progenitor cells and therefore high levels of JAK2V617F in homozygous mutated progenitor cells is required to spontaneously activate EPOR and generate a PV-like phenotype with increased erythrocytes above the limit of normal (Table 4) [25-28]. Sustained high levels of homozygous JAK2V617F mutation during long-term follow-up subsequently does lead to a high spontaneous activation level EPOR and GCSF receptor (GCSFR), which is associated with to extramedullary Myeloid Neoplasia in the Spleen (MNS), splenomegaly and cytokine mediated secondary myelofibrosis (Table 4). The percentage of JAK2V617F positivity and progression from heterozygous to homozygous is strongly correlated with the ability to form spontaneous EEC formation (the hallmark of PV) and with progressive post-PV myelofibrosis (Figure 6) [21,28,29].

Figure 6: Relationship of splenomegaly and MPN disease duration to
the JAK2V617F mutation status of burst forming unit-erythropiesis (BFU-E)
clonal genotypes in 6 ET, 14 PV and 6 MF patients [21]. Progression of
heterozygous JAK2V617F mutation load from about 25% to 100% is seen in
ET, PV and MF during long term follow-up of 10 to 20 years. The mutation
load of combined hetero/homozygous JAK2V617F muated PV in 10 cases of
PV was around 25% to 50% in early stage PV and increases to 80% to 100%
in advanced myeloproliferative stages (splenomegaly) of PV and post-PV
myelofibrosis [21].
Godfrey et al, studied the genotype of individual BFU-E in 29 JAK2V617F mutated ET and 30 JAK2V617F mutated PV patients expressed as percentage (%) of EEC colonies genotyped as homozygous (red), heterozygous (purple) or wild type (white in Figure 7) [30]. All 29 JAK2V617F positive ET patients have heterozygous JAK2 mutated EEC colonies and a low percentage less than 10% homozyous colonies in 9 and 20% in 1 of them. Out of 30 JAK2V617F positive PV patients 8 have heterozygous JAK2 mutated EEC, 13 have homozygous EEC colonies of more than 50% and 7 of less than 50% (Figure 7). To determine whether JAK2V617F-homozygous colonie were part of a single clone or reflected recurrent acquisition of Loss Of Heterozygosity (LOH), breakpoints for chromosome 9p LOH were mapped using fluorescence microsatellite PCR in 576 homozygous mutant colonies from 10 patients (8 PV and 2 ET). Homozygous mutant colonies were absent or present in low percentages in heterozygous ET, but prevalent and common in patients with JAK2V617F-positive PV [30]. In this small number of patients, PV patients harbored a major homozygous-mutant clone that was 8-85 times the size of minor subclones in the same patient. This observation demonstrates that the large numbers of homozygous mutant colonies present in most PV patients do not reflect accumulation of numerous independent subclones but rather the expansion of one dominant clone. The selective expansion of one dominant homozygous subclone is likely to reflect additional cytogenetic [31], genetic or epigenetic alterations in ET, PV and MF patients [32,33]. Such acquired additional epigenetic background biological factors on top of the JAK2, MPL and CALR driver mutations of MPN are associated with impaired prognosis and will become of huge importance for the understanding of differences in biology, prognosis and outcome [34,35].
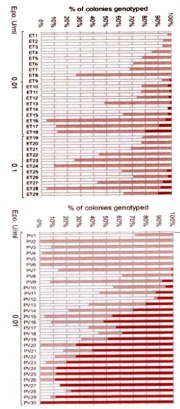
Figure 7: Proportions of JAK2V617F genotypes in BFU-Es from 59 patients
with JAK2-mutated essential thrombocythemia (ET) and polycythemia vera
(PV) [30]. Each vertical bar represents 1 patient, divided according to the
proportion of wild-type, heterozygous, and homozygous-mutant colonies
obtained, with the absolute colony numbers shown above: (wild type white),
heterozygous (purple) homozygous (red). Results EEC colony genotypes are
presented for 29 JAK2V617F-positive ET patients (total 2277 colonies; mean
79 per patient) and for30 JAK2V617F-positive PV patients (total 2287 colonies;
mean 76 colonies per patient) [30].
All 29 JAK2V617F positive ET patients have heterozygous JAK2 mutated
EEC colonies and a low percentage less than 10% homozygous colonies
in 9 and 20% in 1 of them. Out of 30 JAK2V617F positive PV patients 8 have
heterozygous JAK2 mutated EEC, 13 have homozygous EEC colonies of
more than 50% and 7 of less than50%.
Change of 2016 WHO and ECMP into 2019 CLMP Criteria
Using Next Generation Sequencing (NGS), Lundberg et al, found that 28 of 104 (27%) of genes analyzed were mutated in at least 1 (JAK2 or CALR) of the 197MPN patients diagnosed according to 2008 WHO criteria as PV in 94, ET in 69, and PMF in 34 (Figure 8) [35]. The JAK2V617F mutation was recorded in 2008 WHO defined ET, PV and PMF patients and CALR mutation was recorded in ET and PMF patients and not in PV. Seventeen of 69 (25%) ET patients and 11 of 34 (32%) MF patients carried mutations in CALR. After JAK2V617F and CALR, the most frequently observed mutations affected genes implicated in epigenetic regulation (TET2, ASXL1, DNMT3A, EZH2, and IDH1, Figure 8). JAK2V617F mutation was recorded in ET, PV and PMF patients and CALR mutation was recorded in ET and PMF patients and not in PV. Rare mutations include NF1, NFE2, and CUX1. Recurrent somatic mutations were observed in the genes TP53, CBL, MPL, and NRAS. Non recurrent mutations were detected in 16 other genes. The NGS approach also detected copy number alterations, for example, deletions on chromosome. Overall, 20 of 197 patients (10%) had no detectable somatic mutation in any of the genes analyzed (9 ET, 7 PV, and 4 PMF). On top of the JAK2 or CALR mutation one, two or more somatic mutations were found in 65 of197 (33%) patients, which appeared to be of impaired prognostic significance [35].
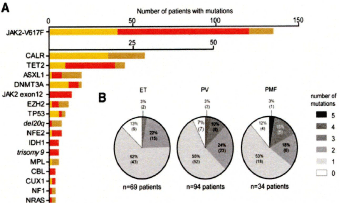
Figure 8: Distribution of somatic mutations in 197 MPN patients from the
study of Lundberg et al [35]. None of 94 (%) PV patients, 17 of 69 (25%) ET
patients and 11 of 34 (32%) MF patients carried mutations in the caleticulin
(CALR) gene. After JAK2V617F and CALR, the most frequently observed
mutations affected genes implicated in epigenetic regulation (TET2, ASXL1,
DNMT3A, EZH2, and IDH1. Rare epigenetic mutations include NF1, NFE2,
and CUX1. Recurrent somatic mutations were observed in the genes TP53,
CBL, MPL, and NRAS. On top of the JAK2 or CALR mutation one, two or
more somatic mutations were found in 65 of 197 (33%) patients, which
appeared to be of impaired prognostic significance [35].
The 2006-2015ECMP classifications defined at least five distinct MPNs caused by the somatic driver mutation JAK2V617F, JAK2 exon 12, CALR and MPL515 which mutually exclude each other whereas bone marrow fibrosis is a second at event in all variant of MPN (Tables 1 to 7). The ECMP classification clearly defined three phenotypes of JAK2V617F mutated ET: norm cellular ET, hypercelluar ET due to increased erythropoiesis (prodromal PV) and ET with hypercellular megakaryocytic-granulocytic myeloproliferation (EMGM or masked PV (Table 1) [6-9]. The updated 2019 CLMP for the diagnosis of PV (Table 2) and subsequent staging of PV distinguished at least 6 stages that has important therapeutic implications (Table 5) [7-9]. Bone marrow cellularity, increased erythropoiesis or granulopoisis and the morphology of pleomorphic megakarocytes are not different in JAK2V617F mutated ET, prodromal PV, masked PV and classical PV (Figure 5). Normocellular ET had stable ET disease without any progression during lifelong follow-up. WHO defined ET patients frequently had a typical hypercellular PV bone marrow picture due to increased erythropoiesis similar as observed in newly diagnosed PV patients (Figures 2 and 6) [7-9,21]. JAK2V617F mutated pure erythrocythemia or idiopathic erythrocytosis according to PVSG criteria presented with a typical PV bone marrow histology and persistant increased erythrocyte counts above 6×1012/L (Figure 4). After correction of haemoglobin and hematocrits to around 0.40 by repeated venasections (Figure 4) the erythrocyte counts remained above 6×1012/L whereas the JAKV617F mutation load increased in this case raised from heterozygous 25% to homozygous 65% after 5 years follow-up.

Figure 4: Clinical course in a case with idiopathic erythrocythemia (IE) or
erythrocy themic polycythemia vera treated with venesections (arrows). The
development of microcytic hypochromic erythrocytes due to iron deficiency
was associated with persistent increased red cell count (above 6x1012/L),
which is diagnostic for PV. Phlebotomy on top of low dose aspirin induces
iron deficiency with microcytic erythrocytes (MCV around 65 fL), normal
values for haemoglobin (Hb) and hematocrit (Ht) and relief of hypervolumic
symtoms.
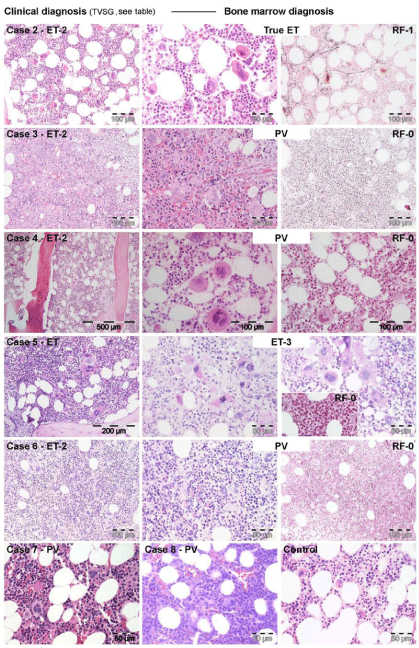
Figure 5: 5: WHO bone marrow features in JAK2V617F+ET cases 2, 3, 4, 5 and 6,
and PV case 7describedby Michiels et al in 2014 [7].
Case 2: Clinically JAK2V617F+ ET 2 (low serum EPO) and a normocellular ET
phenotype 1 bone marrow wit pleomorph small and large megakaryocytes
and reticulin fibers: RF grade 1.
Case 3 and 4: Clinically JAK2V617F+ ET phenotype2 with a trilinear
hypercellular PV bone marrow and RF 0 in case 2 and increased cellularity
due to increased erythropoiesis in case 4, and no increase of reticulin fibers:
RF grade 0.
Case 5: Clinically JAK2V617F+ ET withslightto moderate splenomegaly and
a hyper cellular mega karyocytic granulocytic myelo proliferation (EMGM =
ET phenoype3), with dysmorphic megakaryocytes (not cloud-like) and no
increase of reticulin fibers: RF grade 0 in the bone marrow.
Case 6: Clinically JAK2V617F+ ET phenotype2 with a trilinear hyper cellular PV
bone marrow picture and no increase of reiculine fibers: RF 0.
Case 7: Clinically JAK2V617F+ PV with a 65% hyper cellular ET/PV bone
marrow picture in between “normo cellular ET” and trilinear hyper cellular
(100%) PV picture.
![]()
PV: CMP stage
0
1
2
3
4
5
6
Clinical Diagnosis
Prodromal PV
Erythrocythemia
Early PV
Classical PV
Masked PV
InapparentPV: IPV
Post-PV MF
LAP-score, CD11B
↑
↑
↑
↑
↑/↑↑
↑
variable
EEC
+
+
+
+
+
+
+
Serum EPO
N/↓
N/↓
↓
↓
↓
↓
variable
Erythrocytes x1012/l
<5.8
>5.8
>5.8
>5.8
<5.8
<5.5
↓
Leukocytes x109/l
<12
<12
<or >12
< or->15
>15
N or ↑
>20
Platelets x109/l
>400
,400
< or >400
>400
+1000
N or ↑
variable
CLMP bone
marrow histology
Early PV
Early PV
EMGM
EMGM
EMGM
MG-MF
MF
BM cellularity (%)
Grading RF[44-47]
Grading MF[44-47]
50-80
RF 0-1
MF 0
50-80
RF 0-1
MF 0
60-100
RF 0-1
MF 0
80-100
RF 0/1,
MF 0
80-100
RCF2/3
MF 1/2
60-100
RCF 2/3
MF 1/2
↓
RCF 3/ 4
MF 2/3
Spleen size:
Onechogram
Below MCL
<12-15
0-3
<13
NP
12-15
0-3
12-16
4-6
18->20
>6
16 >20
>6
>20 cm
>8 cm
JAK2V617F� load
Granulocytes %
low
+(++)
low
+(++)
Moderate <50% +
Mod/High
+ / ++
High >50% ++
High
>50% ++
High
>50% ++
Risk stratification
àTherapeutic implications
Low
Low
Low/mod
Intermediate
High
early MF
JAK2 inhibitor
Post-PV MF
First line
Aspirin/Phlebotomy
Second line IFN
versus (HU)
Third line JAK2
inhibitor
Aspirin
Phlebotomy (Phleb)
Aspirin
Phleb
Phleb
Aspirin
Low dose IFN à responsive
Phleb*
Aspirin
IFNà
resistant à HU
IFN
resistant à
HU
or JAK2
inhibitor
IFN
resistent
àJAK2
inhibitor
JAK2
Inhibitor à
Bone marrow
Transplant
*↑ = increased, ↓ = decreased, N = normal, + = present or heterozygous; ++ = homozygous Designed by Michiels 2000-2019
Table 5: Staging of JAK2V617F positive prodromal PV, erythrocythemic PV, classical PV, early MF, inapparent PV, spent phase PV and post-PV myelofibrosis (MF) according to 2019 CLMP criteria related to therapy.
Detection of JAK2V617F mutation and serum EPO measurement have become the first step in the diagnostic work-up of erythrocytosis with erythrocyte counts above the upper limit of normal (>5,6×1012/L) [35-38]. Vannucchi et al, employed quantitative assays for JAK2V617F allele levels in granulocytes in a prospective study of 175 PV patients at time of diagnosis [18]. The JAK2 mutant allele burden could be quantified as 1%-25%, 25% to 50%, 50%-75% and 75%-100% in 57, 50, 34 and 32 PV patient respectively at time of investigation [18]. The burden of JAK2V617F allele was directly correlated with abnormally increased levels of hematocrit, white cell and neutrophil count, LDH and LAP score, spleen size on echogram and with decreased values for serum ferritin, and erythropoietin [18]. The JAK2V617F mutation load correlated with a progressively higher relative risk for aqua genic pruritus, spleen size on echogram, total thrombosis and the need for receiving myeloma suppressive agents (hydroxyurea).
JAK2 exon 12 Mutations is a Distinct PV Discovered by Green’s Team on MPN UK
The UK MPN study group of Green and co-workers discovered JAK2 exon 12 mutations by screening the complete JAK2 gene in JAK2V617F negative cases of classical PV [38]. The frequency of JAK2 exon 12 mutations among all patients with PV is estimated around 3% [39,40]. JAK2 N542-E543del is the most frequent among the different reported exon 12 mutations. The finding of the JAK2 exon 12 mutations in the 5% PV patients negative for JAK2V617F usually present with early stage PV or idiopathic erythrocytosis [Idiopathic Erythrocythemia (IE)= increased red cell mass with normal values for leukocytes and platelets and no palpable spleen]with a favorable outcome and normal life expectancy [39-41]. In JAK2 exon 12 mutated PV homozygous clone were absent or the sizes were small and very likely explain he benign course of the MPN disease. A low percentage of homozygosity for the JAK2 exon 12 mutation was observed in both K539L-type and E543del-type mutations. The relative high proportions of heterozygous mutant colonies were stable over time in 16 patients tested on 2 separate occasions. Pre-treatment bone marrow histology in JAK2 exon 12 mutated PV or IE showed characteristic erythroid hyperplasia with minor and distinct histology changes of the megakaryocyte lineage. JAK2 exon 12 mutated PV were frequently diagnosed as IE with increased hemoglobin hematcrit and red cell mass, low serum EPO, but normal platelet and leukocyte counts, no or palpable spleen. The bone marrow histology is hypercellular predominantly due to erythroid hyperplasia and clusters of large megakaryocytes with hyperploidnuclei [39-41]. Bone marrow pathology of the JAK2 exon 12 PV cases lacked the prominent clustering of large megakaryocytes and revealed a spectrum of small to medium sized megakaryocyte with a predominance of smaller forms with a varying degree of lobation comprising monolobulated and hyperlobulated forms (Figure 9) [41].
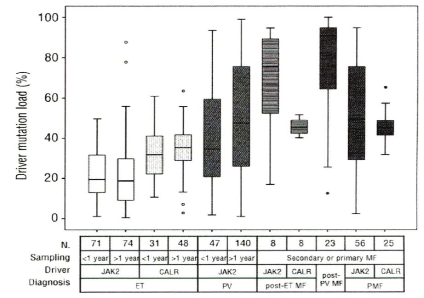
Figure 9: Driver mutation load in WHO defined JAK2V617F ET, CALR ET,
JAK2V617F PV, JAK2V617F post-ET, CALR post-ET, JAK2V617F post-PV PMF and
CALR PMF in the study of Dr Hajnalka Andrikovics et al on Distinct clinical
characteristics of myeloproliferative neoplasms with calreticulin mutations.
Haematologica2014; 99(7): 1184-1190. ET: essential thrombocythemia; PV:
polycythemiavera: MF myelofibrosis: PMF primary myelofibrosis.
CALR Mutated Thrombocythemia is a Distinct MPN Entity Discovered by Kralovics
According to Dameshek (1951) Megakaryocyte Leukemia (ML) is defined by platelet counts around and above 1000x109/L without features of PV in blood and normal erythropoiesis in bone marrow smear and biopsy (Figure 1). The Hannover Bone Marrow classification distinguished three MPD disease entities of ET, PV and hypercellular thrombocythemia related to primary megakaryocytic Myeloproliferation (PMGM, Table 6) without features of PV. The discovery of the Calreticulin (CALR) as the main cause of JAK2/ MPL515 wild type ET and PMF has been immediately identified by Michiels & De Raeve as the driver cause of prefibrotic and fibrotic stages of PMGM without features of PV [8,9]. Kralovics first discovered the Calreticulin (CALR) mutations in a few cases with WHO defined JAK2/MPL wild type ET and PMF patients by next generation sequencing [48]. Dr Kralovics and his team in Vienna Austria subsequently detected Calreticulin (CALR) exon 9 somatic mutations in 78 of 311 (25%) ET patients and in 72 of 203 (35%) MF patients and in none of 382 PV patients [42]. The somatic CALR mutation was not found 45 CML, 73 MDS, 64 chronic myelomonocytic leukemia (CMML) and 24 RARS-T patients except that 3 SF3B1 positive RARS-T patients carried a CALR mutation. Among 1235 ET and MF patients in the Italian-Austrian study, 63.4%, 4.4% and 23.5% carried the JAK2V617F, MPL515 and CALR mutation respectively, and in 8.8% none of these clonal markers (triple negative) was detected [43]. Evolution into MF during very long term follow up was equallly high in CALR mutated ET as in JAK2 mutated PV (about 20% after 20 years follow up). CALR mutated MPN patients had higher platelet counts, normal to low normal hemoglobin and white blood cells counts and a lower incidence of major thrombotic events simple because it lacks PV features [42,43]. The UK study found somatic CALR mutations in 110 of 158 (69%) of JAK2/MPL wild type MPN, including 80 of 112 (70%) ET patients, 18 of 32 (56%) MF patients [44]. CALR exon 9 mutations were found in 26 of 31 (84%) patients with JAK2/MPL wild type MF. CALR exon 9 mutations were absent in all 120 JAK2V617F or MPL mutated patients. CALR mutations were present in 10 of 120 (8%) MDS patients (RA in 5 of 53, RARS in 3 of 27 and RAEB-T in 2 of 27), and in one patient each with CMML and atypical CML. No CALR mutations were found in control samples, lymphoid cancers, solid tumors, or cell lines.
![]()
CLM criteria CALR thrombocythemia
Pathological:P criteria of CALR PMGM
A1 No preceding or allied other subtype of myeloproliferative neoplasm PV, CML, MDS. The main presenting features is pronounced isolated thrombocythemia with platelet count around or above 1000x109/L
A2 Presence of CALR mutation and JAK2 wild typePrimary megakaryocytic granulocytic myeloproliferation (PMGM) and relative or absolute reduction of erythropoiesis and erythroid precursors. Abnormal dense clustering and increase in atypical medium sized, large to giant immature megakaryocytes containing bulbous (cloud-like) hypolobulated nuclei and definitive maturation defects
C Clinical stages of CALR Thrombocythemia
Grading reticulin fibrosis (RF), myelofibrosis (MF) [44-47]
C 1. Early clinical stage: Hb >12g/dL, slight to moderate splenomegaly, thrombocytosis around or above 1000x109/L, normal LAP score
C2. Intermediate clinical stage: slight anemia Hb <12 to >10 g/dL, decreasing platelet count, splenomegaly, increased LDH and definitive tear drop erythrocytes
C3. Advanced stage: anemia Hb <10 g/dL, tear drop erythrocytes, increased LDH, increased CD34+ cells, pronounced splenomegaly, normal or decreased platelet counts, leucocytosis or leukopenia.MF 0 Prefibrotic CALR MGM, no reticulin fibrosis RF 0/1
MF 1 Early fibrotic CALR MGM slight reticulin fibrosis RF 2
MF 2 Fibrotic CALR MGM increase RF grade 3 and slight to moderate collagen fibrosis
MF 3 Advanced fibrotic CALR MGM with collagen fibrosis-osteosclerosisThe combination of A1 + A2 and P1 establishes CALR Thrombocythemia and various clinical stages(C1, C2,C3) related to the degree of secondary myelofibosis (MF)
Table 6: 2019 CLMP criteria for hypercellular ET associated with primary megakaryocytic, granulocytic myeloproliferation (PMGM) caused by calreticulin (CALR) mutations [8,9].
Tefferi et al, retospectively studied 254 evaluable WHO-defined MF patients in whom the JAK2-, MPL- and CALR-mutations were present in 58%, 8.3% and 25% respectively, and 8.7% were triple negative [45]. Median Overall Survival (OS) durations of 83 CALR-, 21 MPL-, and 147 JAK2-mutated MF cases and in 22 triple negative MF cases were 8.2, 4.1, 4.3 and 2.5 years respectively. As compared to JAK2 mutated MF, CALR-mutated MF patients were younger, had higher platelet count, lower leukocyte count, were less anemic with lower DIPSS-plus score. The median overall survival was 2.3 years in 55 CALR-negative/ASLX1-positive, 5.6 years in 126 CALR-negative/ ASXL1-negative MF patients, 7 years in 20 CALR-positive/ASXL1- positive MF patients and 9.6 years in 126 CALR-positive/ASXL1- negative MF patients [45].
Primary Myelofibrosis (PMF) is not a Disease but a Secondary Event in Clonal MPN
The 1975 PVSG criteria for PV, for primary hemorrhagic thrombocythemia and for Primary Myelofibrosis (PMF) are very crude and overlook the early latent and masked stages of MPD [46,47]. In 1977 Silverstein updated the spectrum of PVSG defined Primary Hemorrhagic Thrombocythemia (PTH) versus. PMF [47]. PMF has been defined by the PVSG as a clinic pathological entity not preceded by any other MPD ET, PV, CML, or preleukemia (MDS) [46-51]. PVSG defined PMF is characterized by various degrees of anemia, splenomegaly, leukoerythroblastosis, with tear drop-shaped erythrocytes, and dry tap on BM aspiration. This PVSG definition of PMF is used by the 2001 and 2008 WHO classifications [10-12]. Cytogenetic studies, isozyme markers and gene mutations studies (polymerase chain reaction: PCR) between 1969 and 1981 demonstrated that fibroblast proliferation in ET, PV, and AMM appeared to be polyclonal [28]. This simple indicates that the various degrees of MF (RF and RCF, Table 2) is a reactive process whereas the hematopoietic stem cells appeared to be of clonal origin in each of the chronic myeloproliferative disorders PMF, PV, ET and in CML (reviewed by Michiels et al, 2006 [28]). With the advent of JAK2V617F as the driver cause of trilinear MPNs ET, PV and MF and MPL515 and CALR mutations as the driver causes of two distinct thrombocythemia with various degrees of bone, arrow fibrosis (myelofibrosis: MF), PMF is not a disease anymore but a secondary event in all molecular variants of MPN. At least two kinds of fiber qualities can easily be distinguish by common staining in light microscopy (Table 2): Reticulin Fibrosis (RF) and Collagen Fibrosis (CF). Gommori’s silver staining detects early and course Reticulin Fibers (RF) and do not stain collagen fibers thereby underestimating advanced RCF myelofibrosis grade 2 and 3. Collagen fibers stain with a Mason’s trichrome stains. Silver stain does not distinguish RF from RCF in advanced Myelofibrosis (MF) grade 2 and 3 (Table 2). Consequently both Gommorri’s stain for Reticulin Fibrosis (RF) and trichrome stain for Collagen Fibrosis (CF) are to be used for optimal MF-grading of RF and RCF [48-51]. The evolution of RF into RCF as documented by the combined use of silver and trichrome stains simple means a determinative change from reversible normal reticulin (+RF) into irreversible pathological collagen scarring (+RCF without or with osteosclerose) [48-51]. Clinically, RCF often results in dry tap, when aspiration is attempted. Reticulin fibrosis grade 0/1 and RF with very early Collagen Fibrosis (RCF) usually do occur without real scarring. Bone marrow aspiration in RF without CF usually does not cause the symptom of dry tap. Advanced myelofibrosis (RCF = MF 2 and 3) designates a pronounced increased collagen fibrosis with visuable scarring spotted areas and sometimes with foci or larger areas of atrophic hematopoiesis in the bone marrow in light microscopy.
Bone Marrow Histology of CALR Thrombocythemia: From Dameshek to Georgii & Michiels
From 1994 to 2006, Michiels et al, documented a case of JAK2 wild type ET with a PMGM bone marrow (Figure 10) in a 9-yearold boy (referred to us from Basel, Switzerland) with a platelet count of 1596 to 1946x109/L, no splenomegaly on palpation, white blood differential count (metamyelocytes 0.5%, banded forms 1%, segmented granulocytes 52%, basophiles 2.5%, lymphocytes 35% monocytes 6%), low LAP score, and a hypercellular (80-100%) bone marrow with a predominant prefibrotic primary megakaryocytic and granulocytic Myeloproliferation (PMGM, Table 6), absence of reticulin fibers, loose to dense clustering of large dysmorphic megakaryocytes variable in size with cloud-like hypoploid nuclei. The dysmorphic megakaryocytes show definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei and some disturbances of the nuclear cytoplasmic ratio (Figure 10, arrows), which are not seen in JAK2V617F mutated ET. The 10 years followup from 1994 to 2004 showed normal blood cells counts, absence of the JAK2V617F mutation, no evidence of myelofibrosis, and no splenomegaly on palpation (Figure 10).
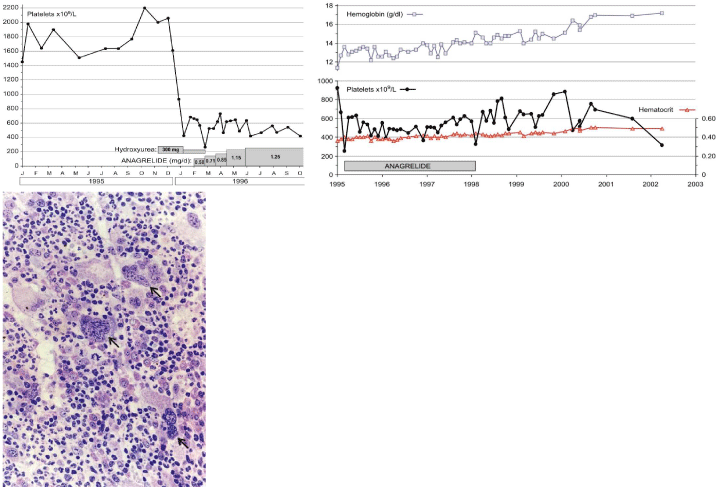
Figure 10: Hemorrhagic thrombocythemia in case of JAK2 wild type hypercellular ET associated with prefibrotic primary megakaryocytic and granulocytic
myeloproliferation (PMGM, colour picture) in the bone marrow by dual myeloproliferation of granulopoiesis and dense clustered enlarged immature dysmorphic
megakaryocytes with bulky (bulbous) hyperchromatic nuclei (arrows), which are never seen in JAK2 wild type MPL515 mutated ET and also not in the prefibrotic
JAK2V617F mutated ET. During long-term follow-up, reduction of platelet count to normal or near normal by treatment with hydroxyurea in 1994 followed by anagrelide
from 1995 to 1998 the bleeding manifestations did not recur. After discontinuation of anagrelide in 1998 the patient remained asymptomatic, the platelet counts
were between 600 and 800x109/L, which normalized after 8 years of follow-up. From 2001 to 2005 haemoglobin and hematocrit reached completely normal values.
Personal observations Dr. Michiels.
The bone marrow histology in 15 consecutive newly diagnosed CALR mutated ET and early MF in 2014/2015 collected by Michiels & De Raeve revealed a typical PMGM picture characterized by dysmorphic megakaryocytes with definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei (Table 6) (manuscript submitted to IJBMR May 30 2019). Representative bone marrow histology findings of typical cases of CALR positive ET (Figure 11) and CALR positive MF (Figure 12) show dense cluster of immature megakaryocytes. The clinical presentation, laboratory and molecular findings has been confirmed in Belgium in 40 of 64 JAK2 wild type MPN (ET or MF) patients [52]. CALR thrombocythemia patients are phenotypically distinct from JAK2V617F mutated ET and prodromal PV cases with regard to clinical and hematological features at presentation and during follow-up.
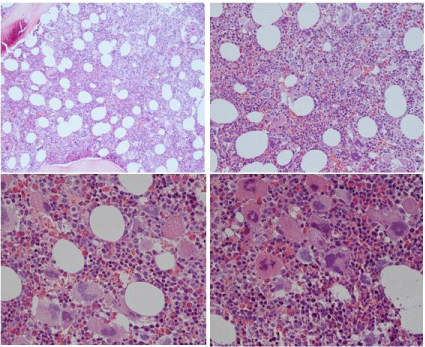
Figure 11: Clinical case of calcireticulin (CALR positive ET who present with
aspirin responsive platelet thrombophilia, normal values for hemoglobin.
Hematocrit and erythrocytes, platelet count of 1352x109/L and slight
plenomegaly (16 cm length diameter on echogram). Bone marrow histology
is hypercellular with relative decrease of erythropoiesis, dense cluster of
immature megakaryocytes with hypolobulated nuclei consistent, and no
increase of reticulin fibrosis consistent with a typical PMGM bone marrow
(Table 7).
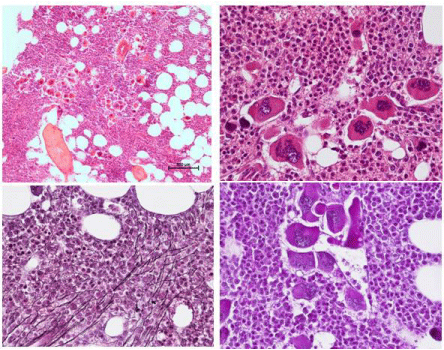
Figure 12: Clinical case of CALR positive myelofibrosis (MF): hemoglobin
11.2 g/dL, hematocrit 0.33, leukocytes 9.2x109/L, platelets 347x109/L, LDH
1393 U/l, and the presence of tear drop erythrocytes, poikolocytosis and
polychromasie in a peripheral blood smear, and hypercellular bone marrow
with relative decrease of erythropoisis, dense cluster of immature mega
karyocytes with hypolobulated nuclei consistent, and reticulin fibrosis grade 2
consistent with a PMGM bone marrow (Table7), clearly distinct from JAK2V617F
mutated ET and PV, and distinct from MPL515 mutated ET (Figure13).
Acquired MPL515 Mutated Normocellular ET: A Rare Distinct Thrombocythemia Entity Discovered by Pikman and Pardani
Three European studies describe MPLW515L and MPLW515K mutations as the cause of acquired clonal ET and myelofibrosis without features of PV [53-55]. Within the JAK2 wild type MPN, the prevalence of the MPL515 mutation as the cause of ET is 3% in the Vannucchi study [77], and 8.5% in the UK studies [54,55]. The clinical presentation in 30 ET patients with acquired MPL515 mutation (9 males and 21 females, age 22-84 (mean 56 years of whom 18 had the W515L and 12 the W515K) was featured by a high incidence of major arterial event in 23%, venous thrombosis in 10%, aspirin responsive micro vessel disturbances in 60%, and major hemorrhage in 7% [53]. The only abnormal laboratory finding in MPL515 mutated ET was increased platelet counts, 956+331 × 109/L in all and slight splenomegaly in 5 (17%). MPL515 mutated ET patients have no clinical, laboratory and bone marrow features of prodromal PV at diagnosis and follow-up, have normal serum EPO, normal ferritin levels, absence of spontaneous Endogenous Erythroid Colonies (EEC). The pretreatment bone marrow at time of diagnosis in a typical case of MPL515 mutated ET is featured by normocellular ET with pronounced megakaryopoiesis with large and giant megakarocytes and no increase of erythropoiesis (Figure 13, Table 7). The comparison of bone marrow histopathology findings in patients with normocellular JAK2V617F mutated ET versus MPL515 mutated ET show significant differences [8]. The megakaryocytes in MPL515 mutated ET are larger with hyperlobulated staghorn-like nuclei as compared to the pleomorphic megakaryocytes morphology in JAK2V617F mutated ET and PV (Figure 13, Table 7). There was local increase of erythropoiesis in areas of loose clustered pleiomorphic megakaryoctyes in JAK2V617F mutated ET, but not in JAK2 wild type PT carrying the MPL515 mutation.
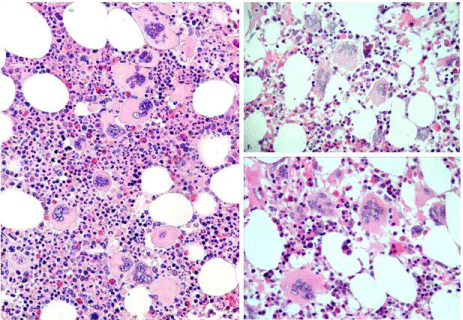
Figure 13: Comparison of JAK2V617F mutated ET versus MPL515 mutated
normocellular ET: 65 years old man with RBC 5.37x1012/L, HB 15.8 g/dL,
MCV 89, WBC 11.95 x109/L, Plts 517x109/L, LDH 600 UI/L and JAK2V617F
mutation (allele burden: 20% on peripheral blood). Diagnosis JAK2V617F
mutated prodromal PV according ECMP (left). 55 years old woman with
MPL515- positive normocellular ET with normal values for hemoglobin,
hematocrit, leukocytes and platelet around 1000x109/L associated with small
medium, large and giant megakaryocytes with staghorn-like hyperlobulated
nuclei (right).
![]()
Clinical. laboratory and molecular: CLM
Bone marrow pathology: P criteria
1. Platelet count >350x109/L and presence of large platelets in blood smear
2. Hemoglobin, haematocrit and erythrocyte count in the normal range
3. Presence of MPL515 mutation and JAK2 wild type
4. Normal serum EPO
5. Normal LAP score and CD11b expression
6. No or slight splenomegaly
7. No leukoerythroblastosis
8. No preceding or allied CML, PV, RAS-T or MDS
P1. Proliferation of large to giant mature megakaryocyte with hyperlobulated, staghorn-like nuclei in a normocellular bone marrow (<60%)
No increase of erythropoiesis, and no increase or immaturity of granulopoiesis or erythropoiesis, No or slight increase in reticulinRF 0/1 Secondary myelofibrosis (MF)
Increased reticulin fibrosis (RF) around dense clustered megakaryocytes in a normocellular bone marrow and reduced erythropoiesis. Follow-up data of RF and (MF) related to splenomegaly in MPL515mutated ET and transltional states to MF are lacking. Grading of reticulin fibrosis (RF) and myelofibrosis (MF) is similar as described for PV
Table 7: 2019 Clinical Laboratory Molecular and Pathological: CLMP criteria for the diagnosis of normocelular MPL515 mutated ET [8,9].
Diagnosis and Treatment of ET, PV and MF in 497 MPN Patients: Dutch Experiences
In 2010, the Dutch MPN Foundation had nearly 700 MPN patient members. A written questionaire was send by the Dutch MPN Foundation to 624 MPN patient members concerning symptoms, treatment, physical mobility, social activity and labour participation. A subgroup of respondents was selected for an additional digital questionaire containing two validated fatigue measurement instruments proposed by Mesa et al, [56,57]. This survey was completed by 450 (response rate 72%) MPN patients and reported in Pur Sang, the MPN magazine of Dutch MPN patients [58]. Since 2000, diagnosis of the MPDs followed the European Clinical Molecular and Pathological (ECMP) and criteria for ET, PV and MF (Table 5). The survey was completed in 2010 by 450 MPN patients (women 56%), resulting in a 72% response rate: ET 39% (n=157), PV 47% (n=213), MF 14% (n=62) [11]. The results of the MPN Questionaires published in PUR SANG anno 2010 were based on 497 filled forms by 271 females (54%) and 212 males (43%), mean age at diagnosis 57 years (range 20 to 84 years) [56]. The 497 MPN patients were diagnosed according to Dutch recommendations in (Table 5) as ET in 181 (36%), PV in 244 (50% of whom 18 as ET/PV), MF in 67 (13%), and MPN unclassifiable in 5 (1%). The primary diagnosis in 115 Dutch and Belgian hospitals was based on specific MPN related complaints in 55%, coincidental (eg routine laboratory investigation for other reasons) in 30% and after disease specific complications had occurred in 15%. Diagnosis of MPN was confirmed by bone marrow aspiration from the sternum in 235 and bone marrow biopsy from the iliac crest in 475 (96%). Red Cell Mass (RCM) measurement to diagnose PV and to distinguish ET from PV was performed in 31%. PCR test for the JAK2V617 mutation was performed in 230 (46%) MPN patients and found positive in74% (ET n=52, PV n=103, MF n=14) and negative in 26%. Sixty percent of ET, 91% of PV and 52% of MF patients were JAK2V617F positive, there by confirming the data in the literature reviewed by Michiels et al, [7,16,29]. After primary diagnosis 144 (25%) MPN patients (ET n= 38, PV n= 49, MF n=27) were referred for a second opinion. The second expert evaluation led to a change in diagnosis in 8% and a change in treatment in 28% (n=29). The second treatment option in 29 (28%) proved to be superior to the intitial treatment. A change of diagnosis during follow-up occurred in 60 MPN patients, from ET into PV in 16 (9% of PV), from PV into MF in 15 (6% of PV), and from ET into MF in 10 (6% of ET).
Based on the Dutch MPN questionaire including 36 questions to answer the top 20 complaints at time of diagnosis in 399 out of 497 (81%) MPN patients is shown in (Table 5). The most frequent complaint is fatigue (81%) equally high in ET, PV and MF patients. Apart from variable severity of fatigue a specific pattern of signs and symptoms could be retrieved by the Dutch MPN questionaire. The signs and symptoms in ET are mainly featured by tingling and prickling sensations in footsoles, handpalms, toes and fingers, cognitive concentration and visual disturbances [59-62]. Itching in PV (58%) and ET (30%) and fatigue were much more prominent in PV. Various degrees of night sweats related to splenomegaly occurred in about half of the MPN patients (Table 5). About one third of MPN patients suffered from bone pain (Table 5). MF patients suffered more frequently from constitutional symptoms of prominent fatigue and night sweats (78%) related to pronounced splenomegaly (Table 5). Treatment in 497 MPN patients was started with low dose aspirin 40 to 80 mg OD (calcium carbasalate Ascal) in 70% and phlebotomy in 42% (mainly PV 91%), hydroxyurea in 29%, and pegylated interferon-alpha 2a in 7%, wait and see in 8% (n=42 of whom 26 with MF) of MPN patients at time of diagnosis [56]. The treatment changed during follow-up in 294 (60%) of MPN patients: ET in 64% (n=115), PV in 59% (n=143) and MF in 49% (n=33). Out of 459 evaluable adverse drug reactions or side effects were recorded in one third (35%) of MPN patients: HU in 41% (n=69), IFN in 28% (n=47) of all side effects. Most frequent side effects of HU were skin and mucocutaneous complaints including dry skin, skin lesions, skin ulcers, itching, skin carcinoma, brittlenails, aphto usulcers and hair loss. Most frequent side effects of IFN were flue-like symptoms, fatigue and mood disturbances. Low dose aspirin induced gastritic complaints in 11% for which treatment with metronazol was usually indicated [59-62].
Out of 497 MPN patients 168 (34%) indicated not to be able any more to participate in their job. Out of 318 MPN patients who still wish to work 18% were completely and 14% partially unable to work as the consequence of MPN disease [56]. As the consequence of their disease, about one fourth of MPN patients are restricted in their activities to walk in 24% (n=117), to bicycle in 22% (n=111), or sports in 24%, (n=117). Out of 497 MPN patients 86% could accept their MPN disease to live with it themselves (78%) by compassion from families and friends in 41% and professional help was given in 12%. In 46 (9%) patient MPN disease was a great suffer and nearly impossible to live with. Collection and analysis of results derived from the Dutch MPN questionaire by the Dutch MPN Foundation is a continuous process.
2019 CLMP Eurasian Classification and Staging of Mpns: Therapeutic Implications
The updated 2019 CLMP classification and staging of patients with MPN will be very helpful in predicting and documenting prospectively the natural history of JAK2V617F mutated ET, prodromal PV and PV patients (Tables 1 and 2), versus. CALR mutated thrombocythemia (Table 6) and MPL515 mutated thrombocythemia (Table 7). The primary involvement of basic researchers, laboratory scientists, molecular biologists and clinicians as well as pathologists are essential to document the natural history at the clinical molecular and bone marrow level to demonstrate that scrutinized and integrated clinical, laboratory, molecular and pathological approaches and intense communications amongst clinicians, molecular biologists and pathologists are warranted in prospective diagnostic and managements studies (Tables 8 and 9, Figures 14 and 15, Dutch experiences). The 2019 CLMP criteria surely will have important implications in choosing proper targeted treatment options for the management and prevention of thrombotic and bleeding complications and serious complications of progressive MPN disease burden in prodromal PV and overt PV (Figure 16). Proper staging of PV in terms of JAK2V617F mutation load, and MPN disease burden including splenomegaly, constitutional symptoms including itching, bone marrow histology and grading of myelofibrosis is of huge importance since it has significant implications for a nonleukemogenic or the least potential leukemogenic treatment options in low, intermediate and high risk PV patients (Table 5) [9.63,64]. A primary rigid venasection regimen aiming at a hematocrit around and below 0.40 seems to us better than the target of ‹0.45 in males and ‹0.42 in females on top of low dose aspirin for the control of activated platelets in MPN (Table 10). According to our extended experiences, this strategy in stage zero, 1 and 2 PV patients (Table 5) will reduce the cumulative incidence of minor and major thrombosis from above 50% to less than 2% per patient/year during long-term follow-up (Table 11) [60,61].
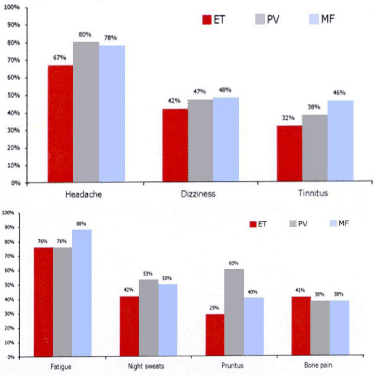
Figure 14: Clinical symptoms of headachewith or without visualdisturbances,
dizziness and tinnitis in the questionaire of 363 Dutch MPN patients subdivided
in 123 (34%) ET, 190 (52%) PV and 50 (14%) patients(upper) 6 and frequency
of fatigue, nights weats, pruritis and bonepain in thequestionaire of 363 Dutch
MPN patients subdivided in 123 (34%) ET, 190 (52%) PV and 50 (14%)
patients [58].
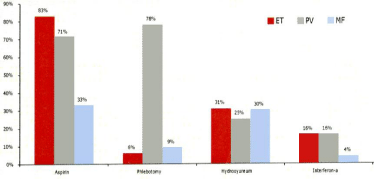
Figure 15: Treatment withaspirin, phlebotomy, hydroxyurea and pegylated interferon in thequestionaire of 363 Dutch MPN patientssubdivided in 123 ET, 190 PV
and 50 MF.
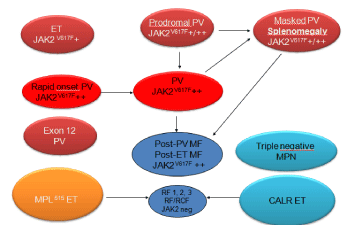
Figure 16: Clinical, Laboratory, Molecular and Pathological (2018 CLMP)
translational states of Myeloproliferative Neoplasms (MPN) Classification of
at least four distinct clonal JAK2V617F, JAK2 exon 12, CALR, MPL mutated
Myeloproliferative Neoplasms (MPN) [69,70].

Figure 17: Discovered and Designed by Michiels et al 2006 [28].
![]()
Patient MPN Diagnosis
PV
ET
MF
Mesa
Michiels
Mesa
Michiels
Mesa
Michiels
Number of patients
405
244
304
181
456
67
Symptomburden
Splenomegaly
42%
36%
24%
22%
56%
78%
Fatigue
85%
81%
72%
80%
84%
85%
Itching
65%
58%
40%
30%
50%
36%
Nightsweats
49%
50%
41%
44%
56%
52%
Bone pain
43%
36%
41%
33%
47%
34%
Therapy
Aspirin
72%
71%
77%
83%
49%
33%
Hydroxyurea
53%
25%
63%
31%
51%
30%
Anagrelide
22%
Near 0%
60%
Low
29%
Near 0%
Interferon
16%
16%
14%
16%
21%
4%
Table 8: Comparative analysis of symptom burden and treatment by diagnosis of the myeloroliferative neoplasms (MPN) as polycythemia vera (PV), essential thrombocythemia (ET) and myelofibrosis (MF) in the study of Mesa et al 2007 [56] and the2010 Dutch MPN study [58] analysed by Michiels.
![]()
Symptoms
Top 15 MPN complaints
MPN
MPN 497
ET 181
PV 244
MF 67
N=497
% of 497
% of 181
% of 244
% of 67
1
Fatigue
399
81
80
81
85
2
Microvascularacra (erythromelalgia)
278
57
61
56
46
3
Cognitivedisturbances
262
53
52
56
45
4
Visual disturbances
249
51
50
52
46
5
Nightsweats
236
48
44
50
52
6
Itching
220
45
30
58
36
7
Dizzines
218
44
44
46
39
8
Bruises, bleedings
211
43
40
45
43
9
Splenomegaly constitutional symptoms
198
40
22
43
78
10
Tinnitus
188
38
38
39
37
11
Migraine
184
37
46
35
22
12
Bone pain
172
35
33
36
34
13
Heartarrythmias
154
31
34
31
24
14
Dysarthria, dyslexia,atypicalTIAs
151
31
31
31
30
15
Hypersensitiveto sounds and noices
149
30
29
32
28
Microvascularacra: Tingling, prickeling sensations, redness, swelling and/or bluish discolouration of foot soles, hand palms, toes and/or fingers: aspirin responsive erythromelalgia [59-62]. Cognitive disturbances of concentration and memory and sudden attacks of unconscienceness [59-62]. Visual disturbances of scintillating scotomas, light flashes, blurred vision, transient monocular blindness, rapid spreading of visual figure disturbances [59,60]. Attacks of migraine-like headaches dysarthria, dyslexia and atypical transient ischemic attacks followed by nausea or vomiting or loss of consciencenous or transient paresis of one extremity [59-62].
Table 9: Top 15 clinical manifestations in 497 patients suffering from myeloproliferative neoplasm (MPN): 181 (36%) essential thrombocythemia (ET) 244 (49%) polycythemia vera (PV), and 67 (14%) myelofibrosis ( MF with hemoglobin<12 g/dL) on the Dutch MPN Questionaire 2010 [58].
![]()
ThromboticRisk
Plateletnumber, age and risk
Treatment option
Plateletnumber 400-1000x109/L
Low doseaspirin 75 mg OD
Low
Age 18 to>80 years
Calcium carbasalate 100 mg OD
No microvascular events, no bleedings
No cardiovascular risk factors
Low to
Platelet number 400-1000x109/L
Low dose aspirin 75 mg OD
Intermediate
Age 18 to>80 years
Calcium carbasalate 100 mg OD
Micro vascular manifestations
Ifplateletcountsabove 1000x109/L and minor or significant bleeding Plateletreductionbyana, IFN or HU
No major thrombosis, no bleedings
No cardiovascular risk factors
Intermediate
Plateletnumber 400-1000x109/L
Low doseaspirin or Calcium carbasalate Consider platelet reduction with anagrelide, IFN or HU
Age 18 to>80 years
Presence of cardiovascular risk factors
High
Plateletnumberabove 1000x109/L
Plateletreduction from above 1000 to below 1000x10/L withana, IFN or HU Aspirin at platelets below 1000x109/L
Age 18 to>80 years
Bleeding at platelets>400x109/L
OD = oncedaily, ana = anagrelide, IFN = pegylatedinterpheron-alpha, HU = hydroxyurea
At platelet count between 1000 and 1500 when on aspirin for microvascular manifestations the risk of bleeding is increased. If bleeding is present reduction at platelet count by anagrelide or IFN from values above to below 1000x109/L is recommended. HU is indicated if case rapid reduction of high to very high platelet count is indicated.� Symptomatic ET with features of PV, splenomegaly and leukocytosis (masked PV or post-ET MF) treatment with low dose IFN is indicated. If non-responsive to IFN or side effects consider hydroxyurea.
Table 10: Dutch treatment recommendations for the use of aspirin and platelet reduction in ET and PV patients with low MPN disease burden: no or slight splenomegaly (‹14 cm on echogram), no or minor increase of Leukocytes (‹15x109/L) no or minor itching, no constitutional symptoms and normal LDH.
The rational for using IFN-a as the first-line treatment option in newly diagnosed JAK2V617F mutated PV patients include its effectiveness to abate constitutional symptoms and to induce a complete remission (Table 5), thereby avoiding phlebotomy, iron deficiency and macrocytosis associated with hydroxyurea [62]. Clinicians will be reluctant to postpone the use of hydroxyurea as long as possible or even lifelong in early stage PV [9,62]. Two studies show IFN-induced Complete Hematological Responses (CHR) within one year, and Major Molecular Responses (MMR) were reached after a follow-up of 2 to 3 years in PV and ET patients [63,64]. The cumulative incidence of MMR was 14% at 2 years and 30% at 4 years follow-up in one prospetive study [63]. Peglyated IFN a-2a (PegasysR) reduced the median JAK2V617F allele burden from 45% to 5% in 37 PV patients in one study [63] and from 64% to 12% in a second study of 79 PV and ET patients [64]. A Complete Molecular Response (CMR) may be reached, which was associated with normalization of bone marrow histology [65]. MPN patients and their physicians should be cautious and attentive since the use of pegylated IFN a-2a or 2b may be associated with significant side effects in about one third of PV patients. Kiladjian and his team of investigators reported in 2015 on the response to pegylated IFN in 31 CALR mutated ET patients during a mean follow-up of 11.8 years [66,67]. A hematological response was achieved in all patients and the median CALR mutational lele burden significantly decreased from 41% at baseline to 26% after treatment. Only 2 CALR ET patients achieved a complete molecular response. In contrast, the percentage of CALR mutation was not significantly modified in CALR ET patients treated with hydroxyurea or aspirin only. The presence of additional mutations (TET2, ASXL1, IDH2 and TP53) was associated with only minor or no molecular IFN responses on CALR mutant clones. MPN patients non-responsive to IFN with progressive myeloproliferative disease, splenomegaly and constitutional symptoms are candidates for myelosuppressive (hydroxyurea) or myeloreductive (JAK2 inhibtors) treatment (Table 5, Figure 16) [62,68-70]. Myelofibrosis transformation of thrombocythemia in MPN of various molecular etiology is a secondary event in MPN and has to be distinguished from the expansion of driver mutation driven clonal MPN (Figure 16) [68-70], on top of germline and epigenetic modifying factors that may result in additional cytogenetic, genetic lesions [31,32]. Such additional, acquired background biological factors on top of the JAK2, MPL and CALR driver causes of MPN will become of huge importance for the understanding of differences in prognosis, evolution and treatment outcome of thrombocythemia and polycythemia patients.
Phlebotomy was the first line treatment in 6% of ET, 78% of PV and 9% of MF. Because of advanced or symptomatic MPN disease 31% of ET, 29% of PV and 30% of MF were on treatment with hydroxyurea and 16% of ET and PV and 4% of MF were on treatment with peglyated interferon (Pegasys R). In the 2007 study of Mesa et al 50 to 60% of ET and PV patients were on hydroxyurea therapy (Table 3) [7]. In the Italian study of Vannucchi et al, [21], a total of 214 patients were treated with phlebotomy, 58% of 219 PV and 4% of 257 ET patients and myelosuppressive chemotherapy with hydroxyurea was administered in 59% of 219 PV and 48% of 257 ET patients. The 20% difference of HU use of 50% of USA MPN patients, 59%/48% in Italian ET/PV patients versus 30% of Dutch ET/ PV patients) can readily be ascribed to significant differences in the USA/Italian WHO guidelines versus the Dutch ECMP guidelines for MPN disease in ET and PV patients [13-15]. In view of the chronic and progressive nature of MPN disease during long term follow-up justify the huge importance to come up with large scale Prospective Unmet Need (PUN) crosssectional studies and PUN studies in newly diagnosed MPN patients in whom the effect of treatment with aspirin, phlebotomy/aspirin, pegylated interferon, anagrelide, hydroxyurea and JAK2 inhibitors in ET, PV and MF patients of various molecular etiology in well-defined MPN patients. The treatment efficacy should not only be defined as the capacity to decrease thrombosis and hemorrhages (Table 11), but should also include the capacity to reduce mutational lele burden and MPN disease burden (Table 5) and the effects on quality of life and work paticipation as well. Treatment options including IFN HU and Ruxilitinib should be evaluated not only directed towards clear indications and efficacy of the nonleukemogenic agents in particular, but that the effects on fatigue, quality of life and labour participation should be incorporated as well.
References
- Dameshek W, Henstell HH. The diagnosis of polycythemia. Ann Intern Med. 1940; 13: 1360-87.
- Dameshek W. Physiopathology and corse of polycythemia vera as related to therapy. JAM. 1950; 142: 790-7.
- Michiels JJ. Physiopathology, etiologic factors, diagnosis and course of polycythemia vera as related to therapy according to William Dameshek 1940-1950. Turkish J Hematol. 2013; 30: 102-110.
- Dameshek W. The treatment of Polycythemia. Blood. 1946; 1: 256.
- Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951; 6: 372-375.
- Michiels JJ, Berneman Z, Schroyens W, De Raeve H. PVSG and WHO vs European Clinical, Molecular and Pathological (ECMP) criteria for prefibrotic myeloproliferative neoplasms. World J Hematol. 2013; 2: 71-88.
- Michiels JJ, Ten Kate F, Lam KH, Schroyens W, Berneman Z, De Raeve H. The European Clinical, Molecular and Pathological (ECMP) criteria and the 2007/2008 revision of the world Health Organization fort he diagnosis, classification and staging of prefibrotic myeloprloiferativeneoplasmscarryigthe JAK2V617F mutation. Turk J Hematol. 2014; 31: 239-254.
- Michiels JJ, Berneman Z,Schroyens W, De Raeve H. Changing concepts of diagnostic criteria of myeloproliferative disorders and the molecularetiology and classification of myeloproliferative neoplasms: From Dameshek 1950 to Vainchenker 2005 and beyond. Acta Haematol. 2015; 133: 71-86.
- Michiels JJ, Valster F, Wielenga J, Schelfout K, De Raeve H. European vs 2015 World Health Organization clinical molecular and pathological classification of myeloproliferative neoplasms. World J Hematol. 2015; 4: 16- 53.
- 2001 WHO classification of the Chronic Myeloproliferative Diseases (CMPD) polycythemiavera, chronicidiopathic myelofibrosis essential thrombocythemia and cMPD unclassifiable. In: Jaffe SS, Harris NL, Stern A, Vardiman JW eds. WHO classification of Tumours of haematopoiesis and lymphoid tissues. Lyon, France IARC. 2001: 31-42.
- Tefferi A, Thiele J. Orazi A. Proposals and rationale forrevision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocy themia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007; 110: 1092-1097.
- Swerd low SH, Campo E, Harris NL. WHO criteria for polycthemiavera, primary myelofibrosis and essential thrombocy themia. WHO Classification of Tumours of Haematopoietic and Lympoid Tissues. Lyon France IARC Press. 2008; 40-50.
- James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. A uniqueclonal JAK2 mutation leading to constitutive signalling causes polycythemia vera. Nature. 2005; 434: 1144-1148.
- Vainchenker W, Constantinescu SN. A unique activating mutation in JAK2 V617F is at the origin of polycythemia vera and allows a new classification of myeloproliferative diseases. Hematology (Am Soc Hematol Educ Progr). 2005; 195-200.
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase in human myeloproliferative disorders. Lancet. 2005; 365: 1054-1061.
- Michiels JJ, Berneman Z, Van Bockstaele D, Van Der Planken M, De Raeve H, Schroyens W. Clinical and laboratory features, pathobiology of plateletmediated thrombosis and bleeding complications and the molecularetiology of essential throm bocythemia and polycythemiavera: the rapeuticimplications. Sem Thromb Hemostas. 2006; 32: 174-207.
- Antonioli E, Guglielmelli P, Pancrazzi A, Bogani C, Verrucci M, Ponziani V, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia. 2005; 19: 1847-49.
- Vannucchi AM, Antonioli E, Guglielmelli P, Longo G, Pancrazzi A, Ponziani V, et al. Prospective identification of high-risk polycythemia vera patients based on JAK2V617F allele burden. Leukemia. 2007; 21: 1952-1959.
- Passamonti F, Rumi E, Pietra D, DellaPorta MG, Boveri E, Pascutto C, et al. Relation between JAK2 V617F mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood. 2006; 107: 3676-3682.
- Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F JAK2 mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006; 108: 2435-2437.
- Moliterno AR, Williams DM, Isaacs MA, Spivak JL. Phenotypic variability with in the JAK2V617F-positive MPD: roles of progenitor cell and neutrophil allele burden. Exp Hematol. 2008; 36: 1480-1486.
- Gale RE, Allen AJR, Nash MJ, Linch DC. Log-term serial analysis of X-chromo some in activation patterns andJAK2 V617F mutant levels in patients with essential thrombocythemia show that minor mutant-positive clones can remain stable for many years. Blood 2007; 109: 1241-1243.
- Michiels JJ, Ten Kate FWJ, Vuzevski VD, Abels J. Histopathology of erythromelalgia in thrombocythemia. Histopathology 1984; 8: 669-678.
- Michiels JJ, Abels J, Steketee J, van Vliet HH, Vuzebvski VD. Erythromelalgia caused by platelet-mediated arteriolar in Flammarion and thrombosis in thrombocythemia. Ann Intern Med. 1985; 102: 466-471.
- Michiels JJ, Berneman Z, Schroyens W, Koudstaal PJ, Lindemans J, van Vliet HH. Platelet-mediated thrombotic complications in patients with ET: reversal by aspirin, platelet reduction, and not by Coumadin. Blood CellsMol Dis. 2006; 36: 199-205.
- Delhommeau F, Pisani DF, James C, Casadevall N, Constatinescu S, Vainchenker W. Oncogenic mechanism in myeloproliferative disorders. Cell Mol Life Sci. 2006; 63: 2939-2953.
- Villeval JL, James C, Pisani DF, Casadevall N, Vainchenker W. New insights into the pathogenesis of JAK2V617F-positive myeloproliferative disorders and consequences for the management of patients. Sem Thromb Hemostas. 2006; 32: 341-351.
- Passamonti F, Rumi E, Pietra D et al. Relation between JAK2 V617F mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood. 2006; 107: 3676- 3682.
- Michiels JJ, De Raeve H, Berneman Z, Van Bockstaele D, Hebeda K, Lam K, Schroyens W. The 2001 world health organization and updated Europe an clinical and pathological criteria for the diagnosis classification and staging of the Philadelphia-negative chronic myeloproliferative disorders. Sem Thromb Hemostas. 2006; 32: 307-340.
- GodfreyAL, Chen E, Pagano F, Ortmann CA, Silber Y, Bellosillo B, et al. JAK2V617F homozygosity arises commonly and recurrently in PV and ET, but PV is characterized by expansion of a dominant homozygoussubc lone. Blood. 2012; 120: 2704-2707.
- Bench AJ, Nacheva EP, Champion KM, Green AR. Molecular genetics and cytogenetics of myeloproliferative disorders. Bailiere’dclin Haematol. 1998; 11: 819-848.
- Gotlib J. JAK2 inhibition in the myeloproliferative neoplasms: lessons learned from the bench and bedside. Heamatology ASH Education Book. 2013; 529- 537.
- Vannucchi A, Lasho TL, GuglielmelliP, Biamonte F, Pardani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013: 27: 1861-1869.
- Guglielmelli P, Lasho TL, Rotunno G, JScore J, Mannarelli C, Pancrazzi A, et al. The number of prognostic ally detrimental mutations and prognosis in primary myelofibrosis: an inter national study of 797 patients. Leukemia. 2014; 28: 1494-1500.
- Lundberg P, Karov A, Nienbold R, Looser R, Hao-Shen H, Nissen A, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014; 123: 2220-2228.
- James C, Delhommeau F, Marzac C, Teyssandier I, Le Couédic JP, Giraudier, et al. Detection of JAK2 V617F as a first intention diagnostic test for erythrocytosis. Leukemia. 2006; 20: 350-353.
- Tefferi A, Pardanani A. Mutation screening for JAK2V617F: when to order the test and how to interpret the results. Leuk Res. 2006; 108 :3472-3476.
- Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007; 356: 459-460.
- Pardani A, Lasho TL, Finke C, Hanson CA, Tefferi A. Prevalence and clinic opathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia. 2007; 21: 1960-1963.
- Lakey MA, Pardani A, Hoyer JD, Nguyen PL, Lasho TL, Tefferi A, et al. Bone marrow morphologic features in polycythemia vera with JAK2 exon 12 mutations. Am J Clin Pathol. 2010; 133: 942-948.
- Laszlo J. Myeloproliferative Disorders (MPD): myelofibrosis, myelosclerosis, extramedullary hematopoiesis, undifferentiated MPD and hemorrhagic thrombocythemia. Semin Hematol. 1975; 12: 409-432.
- Klampf T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013; 369: 2379-2387.
- Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, Milosevic JD, et al. Jak2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcome. Blood. 2014; 123: 1544-1551.
- Nangalia J, Massie CE, Baxter J, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N Engl J Med. 2013; 369: 2391-2405.
- Tefferi A, Lasho TL, Finke CM, Knudson RA, Ketterling R, Hanson CH, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014; 28: 1472-1477.
- Berlin NI. Diagnosis and classification of the polycythemias. Sem Hematol. 1975; 12: 339-351.
- Silverstein MN. Myeloproliferativw diseases. Hematology review. Postgraduate Medicine. 1977; 61: 206-201.
- Georgii A, Vykoupil KF, Buhr T, Choritz H, Döhler U, Kaloutsi V, et al. Chronic myeloproliferative disorders in bone marrow biopsies. Path Res Pract. 1990; 186: 3-27.
- Georgii A, BuhrT, Buesche G, Kreft A, Choritz H. Classification and staging of Ph-negative myeloproliferative disorders by histopathology from bone marrow biopsies. Leuk Lymphoma. 1996; 22: 15-29.
- Thiele J, Kvasnicka HM, Facchetti F, Franco V, Van Der Walt J Orazi A. European consensus for grading bone marrow fibrosis and assessment of cellularity in myeloproliferative disorders. Haematologica. 2005; 90: 1128- 1132.
- https://www.ncbi.nlm.nih.gov/pubmed/17885079
- Al Assaf C, Van Obbergh F, Billiet J, Lierman E, Devos T, Graux C, et al. Analysis of phenotype and outcome in essential thrombocythemia with CALR or JAK2 mutations. Haematologica. 2015; 11: 893-837.
- Vannucchi AM, Antonioli E, Guglielmelli P, Pancrazzi A, Guerini V, BarosiG, et al. Characteristics and clinical correlates of MPL515W>L/K mutation in essential thrombocythemia. Blood. 2008; 112: 844-847.
- Beer PA, Campbell PJ, Scott LM. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008; 112: 141-149.
- Jones AV, Campbell PJ, Beer PA, Schnittger, Vannucchi AM, Zoi K, et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood. 2010; 115: 4517-4523.
- Mesa RA, Niblack J, Wadleigh M, Verstovsek S, Camoriano J, Barnes S, et al. The burden of fatigue and quality of life in myeloproliferativedisorders (MPDs). Cancer. 2007; 109: 68-76.
- Mendoza TR, Wang XS, Cleeland CS, Morrissey M, Johnson BA, Wendt JK, et al. The rapid assessment of fatigue severity in cancer patients. Cancer. 1999; 85: 1186-1196.
- Commandeur S. 500 MPD-ers onder de loep. PRIMEUR: Resulaten MPD Enquete anno 2010. PUR SANG, het mpd-magazine. 2010; 7: 12-15.
- Michiels JJ, Koudstaal PJ, Mulder AH, van Vliet HH. Transient neurologic and ocular manifestations in primary thrombocythemia. Neurology. 1993; 43: 1107-1110.
- Michiels JJ, Van Genderen PJJ, Jansen PHP, Koudstaal PJ. Atypical transient ischemic attacks in thrombocythemia of various myeloproliferative disorders. Leuk Lymphoma. 1996; 22: 65-70.
- Michiels JJ, Ten kate FWJ, Koudstaal PJ, Van Genderen PJJ. Aspirin responsive platelet thrombophilia in essential thrombocythemia and polycythemia vera. World J Hematol. 2013; 2: 20-43.
- Michiels JJ. Myeloproliferative and thrombotic burden and treatment outcome of thrombocythemia and polycythemia patients. World J Crit Care Med. 2015; 4: 230-239.
- Kiladjian JJ, Cassinat B, Turlure P, Cambier N, Roussel M, Bellucci S, et al. High molecular response rate of polycythemia vera treated with peglyated interpheron-alpha-2a. Blood. 2006; 108: 1281.
- Quintas-Cardama A, Kantarjian H, ManshouriT, Luthra R, Estrov Z, Pierce S, et al. Peglyated interferon alfa-2a yields high rates of hematological and molecular response in patients with advanced essential thrombocthemia and polycythemia vera. J Clin Oncol. 2009; 27: 5418-5424.
- Larssen TS, Moeller MB, de Striker K, Nørgaard P, Samuelsson J, Marcher C, et al. Minimal residual disease and normalization of the bone marrow after long term treatment with alfa-interferon2b in polycythemia vera. A report on molecular responses in seven patients in sustained complete haematological remission. Hematology. 2009; 14: 331-334.
- Cassinat B, verger E, Kiladjian JJ. Interferon alpha therapy in CALR-mutated essential thrombocythemia. N Eng J Med. 2014; 371: 188-189.
- Verger E, Cassinat B, Chauveau A, Dosquet C, Giradier S, Schlagter MH, et al. Clinical and molecular response to interferon-alpha therapy in essential thrombocythemia patients with CALR mutations. Blood. 2015; 126: 2585- 2691.
- Michiels JJ, De Rave H, Valster F, Potters V, Kim Y, Kim M. Extension of 2016 World Health Organization (WHO) classification and a new set of clinical, laboratory, molecular and pathological criteria forthe diagnosis of myeloproliferative neoplasms: from Dameshek to Vainchenker, Green and Kralovics. EMJ. 2017; 2: 72-81.
- Michiels JJ, Berneman Z, Gadisseur A, Raeve HD, Schroyens W, Potter V, et al. Myelofibrosis is a Secondary Event in JAK2 Trilinear Myeloproliferative Neoplasm (MPN) and in CALR and MPL Thrombocythemia: Implications for Novel Treatment Options of Prefibrotic MPN. J Hematol Thromembolic Dis. 2017.
- De Raeve H, Michiels JJ, Valster F, Potters V, Kim Y, Kim M. Novel Clinical, Laboratory, Molecular and Pathological (2018 CLMP) Criteria for the Differential Diagnosis of three Distinct JAK2, CALR and MPL Mutated Myeloproliferative Neoplasms: The Role of Driver Mutation Analysis and Bone Marrow Histology. Int J Cancer Res Ther. 2018; 3: 1-12.
Citation: Michiels JJ, De Raeve H, Popov VM, Trevet M and Trifa A. Change of PVSG-WHO Into The European Clinical Laboratory Molecular and Pathological (2019 CLMP) Criteria for Classification and Staging of JAK2, MPL and CALR Mutated Myeloproliferative Neoplasms: Bone Marrow Characteristics from Dameshek to Georgii, Thiele & Michiels. Ann Hematol Oncol. 2019; 6(8): 1262.