
Case Report
Ann Hematol Oncol. 2019; 6(8): 1264.
Myeloma or Lymphoma: That is the Question? Myeloma Relapsing as an Angio-Immunoblastic T Cell Lymphoma
Chehade R1*, Mangel J1,2, Louzada M1,2, Shepherd J3 and Deotare U1,2
1Department of Medicine, Schulich School of Medicine and Dentistry, University of Western Ontario, Canada
2Division of Hematology, Department of Medicine, Schulich School of Medicine and Dentistry, University of Western Ontario, Canada
3Department of Pathology and Laboratory Medicine, Schulich School of Medicine and Dentistry, University of Western Ontario, Canada
*Corresponding author: Chehade R, Department of Medicine, Schulich School of Medicine and Dentistry, University of Western Ontario, Canada, 1151 Richmond St, London, ON N6A 5A5, Canada
Received: June 05, 2019; Accepted: July 17, 2019; Published: July 24, 2019
Abstract
Angio Immunoblastic T Cell Lymphoma (AITL) is a clinically aggressive type of Peripheral T cell Lymphoma derived from T follicular helper cells. A rare malignancy with poor overall survival, AITL is often accompanied by polyclonal or monoclonal proliferation of B lymphocytes, however, AITL is rarely associated with plasma cell proliferation. Here we describe a new diagnosis of AITL developing in a patient previously treated for multiple myeloma. This highlights a role for circulating plasma cells and interaction with T follicular helper cells in AITL tumorigenesis. To the best of our knowledge, this is the first report of newly diagnosed AITL in a patient with a previous history of multiple myeloma.
Keywords: Angio immunoblastic T cell lymphoma; Multiple myeloma
Abbreviations
AITL: Angio Immunoblastic T Cell Lymphoma; MM: Multiple Myeloma; TFH: T Follicular Helper Cell; VMP: Lite Bortezomib, Melphalan, Dexamethasone; CT: Computed Tomography; FISH: Fluorescence In Situ Hybridization; P53: Protein 53; LDH: Lactate Dehydrogenase; CD: Cluster of Differentiation; BCL2: B-Cell Lymphoma 2; TET2: Tet Methylcytosine Dioxygenase 2; DNMT3A: DNA Methyltransferase 3A; IDH2; Isocitrate Dehydrogenase 2; PLCG1: Phospholipase C Gamma 1; RHOA: Ras Homolog Family Member A; NOTCH1; Notch Homolog 1, Translocation-Associated; M: Male; F: Female; LN: Lymph Node; PB: Peripheral Blood; BM: Bone Marrow; H&E: Hematoxylin and Eosin; EBV: Epstein Bar Virus; EBER: EBV Encoded RNA
Case Report
A 76-year-old Caucasian man presented to the emergency department with confusion, on a background of several weeks of generalized fatigue with fevers and night sweats. He was noted to have significant cervical and supraclavicular lymphadenopathy, as well as hepatosplenomegaly. Serum LDH level was elevated at 248 U/L. Computed Tomography (CT) scans of chest, abdomen and pelvis revealed extensive axillary, thoracic, retroperitoneal and pelvic lymphadenopathy in addition to previously noted bony lytic lesions.
The patient had a past medical history of IgA lambda multiple myeloma with high-risk cytogenetics, including 17p deletion. He initially presented with mild anemia identified on routine blood work by his family doctor in October 2010. The original investigation involved a negative colonoscopy, and in later investigations, he had serum protein electrophoresis which showed significant IgA monoclonal peak. The patient then underwent a bone marrow aspiration and biopsy, which showed 40 to 50% plasma cell infiltration. The bone marrow flow cytometry identified a 6% plasma cell population that was lambda light chain restricted. FISH identified a P53 mutation, detected in 18.5% of the cells. In December 2010, the patient had noticed quite significant height loss associated with severe thoracic back pain. Skeletal survey identified multiple thoracic and lumbar compression fractures, as well as multiple areas of bony lytic lesions in the pubic rami, innominate bones, proximal femur, and proximal right humerus, as well as diffuse osteopenia.
He received nine cycles of bortezomib, melphalan and dexamethasone (VMP Lite Chemotherapy) as initial therapy, requiring dose reductions because of neuropathy in the toes, yet attaining near complete response. He showed evidence of biochemical progression in October 2013 as his IgA had increased up to 12.2 with a monoclonal peak measuring at 3.58 g/L and worsening kidney function. He received fifty-two cycles of bortezomib and dexamethasone until November 2016, as a part of a clinical trial and achieved complete hematological response. He was followed up regularly and was doing well until September 2017 when he was found to have elevated lambda light chain levels and creatinine; later in October, he presented to the hospital with confusion.
After admission to hospital, he underwent an open excisional biopsy of a cervical lymph node as well as a bone marrow biopsy. The lymph node (Figures 1D and 1E) was diffusely effaced by a mass-like proliferation of polymorphous lymphocytes, eosinophils, plasma cells, Hodgkin Reed Sternberg-like cells and occasional immunoblasts with proliferation of high endothelial cells toward the periphery. Immunohistochemistry showed the lymphoid infiltrate to be positive for CD3, CD5, CD7, with dominance of CD4 over CD8 expression, and CD10 (Figure 2) and BCL2 negative. There were scattered EBER positive CD20 positive B lymphocytes (Figure 2). The bone marrow biopsy (Figures 1A, 1B and 1C) showed increased cellularity of 60%, with two abnormal population groups noted: (a) a nodular interstitial and paratrabecular lymphoid infiltrate, composed predominantly of small lymphocytes, eosinophils and intermediate sized lymphocytes with atypical angulated nuclei and moderate amount of pale cytoplasm, as well as scattered large atypical lymphoid cells containing irregular nuclei with vesicular chromatin and prominent nucleoli; and (b) few paratrabecular plasma cell aggregates with atypical features, including large, binucleated, dutcher bodies. The plasma cells were less than 10%, CD56 positive and lambda light chain restricted. The diagnosis was peripheral T cell lymphoma, consistent with AITL. There were minor residual clonal plasma cells in the bone marrow.
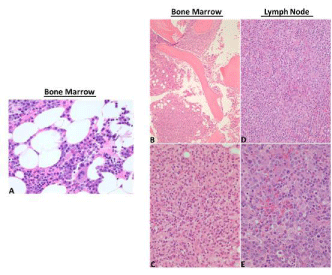
Figure 1: Left panel: A. Hematoxylin and Eosin (H&E) stain on initial bone
marrow biopsy diagnosis of multiple myeloma, showing plasma cell infiltration
(40x). Right panel: B, C. Hematoxylin and Eosin (H&E) stained bone marrow,
and D, E. lymph node biopsies. Bone marrow biopsy, low-power view B
(4x) and high-power view C (40x), reflects architectural changes in AITL
showing proliferation of two abnormal populations, firstly a nodular interstitial
and paratrabecular lymphoid infiltrate composed predominantly of small
lymphocytes, eosinophils, intermediate-sized lymphocytes and scattered
large atypical lymphoid cells, and secondly, some paratrabecular plasma cell
aggregates with atypical features. D, E. Lymph node biopsy, low-power view
D (10x) and high-power view E (40x), showing polymorphous proliferation of
lymphocytes, eosinophils, plasma cells, Hodgkin Reed Sternberg-like cells
and occasional immunoblasts, with proliferation of high endothelial venules.
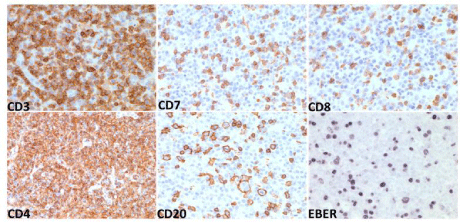
Figure 2: Immunohistochemistry on lymph node biopsy, showing T lymphocyte infiltrates positive for CD3 (also CD5, not shown), CD7, CD8, and CD4, and
scattered CD20 positive B lymphocytes that are also EBV positive as depicted by EBV encoded RNA (EBER).
Intravenous steroids were begun as initial treatment for his lymphoma, but he did not improve. He had a prolonged course at the hospital with increasing oxygen requirements, acute kidney injury and volume overload requiring vasopressor support, intubation and eventual tracheostomy. He subsequently developed systemic candidemia requiring antifungal treatment, and had a positive bronchoalveolar lavage for Corynebacterium striatum treated with vancomycin. His clinical status continued to decline, and he died before receiving more definitive chemotherapy for his lymphoma.
Discussion
Angio immunoblastic T cell lymphoma is a rare malignancy accounting for 2% of all non-Hodgkin lymphomas and 20% of peripheral T cell lymphomas [1]. Recently the World Health Organization recognized AITL under the category of nodal T follicular helper cells (TFH) [1]. AITL usually presents with advanced stage disease in the 6th or 7th decade with a male predominance [1]. Common signs and symptoms include lymphadenopathy, B symptoms, splenomegaly and rash. Bone marrow involvement increases with disease progression [1]. Characteristic laboratory abnormalities at presentation include anemia, thrombocytopenia, elevated lactate dehydrogenase, elevated beta 2-microglobulin, hypergammaglobulinemia, positive direct antiglobulin test, cryoglobulinemia, and cold agglutinins [1]. The prognosis is poor with a 5 year overall survival in most studies of less than 50% [1].
In addition to T follicular helper cells, AITL is characterized by infiltration of a number of accessory cells whose role in pathogenesis remains to be fully determined. There is marked proliferation of endothelial venules with follicular dendritic cells(FDC) around the venules, as well as diffuse polymorphic infiltrates, including the expansion of EBV positive/negative B cells [1-4]. B cells look like Reed- Sternberg cells and this is why AITL may be initially mistaken for Hodgkin lymphoma [3]. Understanding the relationship between TFH cells and B cells in AITL is yet to be determined. Normally, the role of TFH is to help germinal center B cells to differentiate into antibody secreting plasma cells or memory B cells. Therefore, aberrant TFH cells might preferentially select for aberrant polyclonal or clonal plasma cells. Circulating plasma cells and plasmacytoid lymphocytes have been occasionally described in AITL patients [5-10]. There are a handful of reports demonstrating the association between polyclonal plasmacytic proliferation and AITL [5]. These cases were suggested to be associated with EBV infection or due to increased release of cytokines that promote proliferation of plasma cell progenitors [11]. Others have reported on AITL and B cell lymphomas occurring simultaneously [12]; or concurrent diagnosis of AITL and multiple myeloma [7,13] (Table 1). However, there are no prior reported cases of AITL developing years after a diagnosis of multiple myeloma.
![]()
Study
Age/Sex
Type of plasmacytosis with AITL/ Location
Sakai et al, [9]
73/M
Polyclonal, PB and BM
Yamane et al, [10]
63/M
Polyclonal, PB and BM
Suarez et al, [8]
77/F
Monoclonal, cutaneous skin lesion and LN
Zettle et al, [3]
61/M
Clonal (Plasmacytoma), LN
Balague et al, [3]
70/F
56/M
Clonal, LN
Huppmann et al, [4]
60/F
Clonal, LN
Ahsanuddin et al, [2]
76/F
43/F
60/M
Polyclonal, PB and BM
Xu et al, [7]
80/M
Monoclonal, LN and BM
With Plasma Cell Myeloma
Jang et al, [13]
73/M
Monoclonal, LN and BM
With Plasma cell Myeloma
Mitsuhashi et al, [6]
70/M
Polyclonal, PB and BM
Singh et al, [11]
54 /M
54/M
Polyclonal, LN and BM
Polyclonal, LN and BM
Mukherjee et al, [12]
Five M
46�54
Polyclonal, PB, LN and BM
Nagoshi et al, [5]
75/F
68/M
60/M
Polyclonal, PB and BM
Polyclonal, PB and BM
Polyclonal, PB and BM
M=male, F= female, PB= peripheral blood, BM= bone marrow, LN= lymph node.
Table 1: Cases of AITL with MM.
One postulated mechanism explaining the co-occurrence of AITL with plasma cell dyscrasias is that the proliferation of aberrant TFH selects for certain pro-tumorigenic B cell clones and subsequent development of multiple myeloma [1,14,15]. An interesting feature of this case is the development of AITL approximately 3 years following the treatment of multiple myeloma.
A recent genetic analysis of peripheral T-cell lymphomas identified five of the genes most frequently mutated in AITL, few of which are also expressed early on in B cells [14,15] (Figure 3). The genes include epigenetic modifiers, such as TET2, DNMT3A, IDH2, PLCG1, as well as RHOA, a member of the Rho family of GTPases [14,15]. TET2 and DNMT3A mutations were identified in premalignant TFH cells as well as cancer cells [14,15]. RHOA and IDH2 mutations were found only in cancer cells, suggesting that these mutations are acquired in the later processes of AITL development [14,15]. Interestingly, TET2 and DNMT3A mutations were found in tumor infiltrating B cells [14,15]. NOTCH1 mutations, known to be unique to B cells, have also been found in AITL [14,15]. In tracing the etiology of AITL, subsequent to TET2 and DNMT3A mutations, other oncogenic events such as RHOA mutations in TFH cells, microenvironment and cytokine activation, as well as NOTCH1 mutations in B cells, accumulate leading to the development of AITL. The data suggests that the accumulation of somatic mutations might select for clonal expansion of aberrant hematopoietic stem cells which can give rise to multiple cancers sequentially or simultaneously.
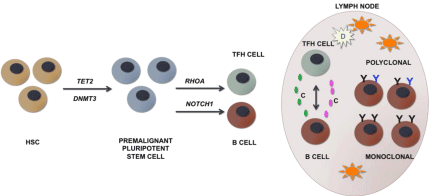
Figure 3: Model of AITL tumorigenesis. (Adapted from Cortés et al, and Fujisawa et al, [14,15]).
Mutations in TET2 and/or DNMT3A are the earliest oncogenic events occurring in undifferentiated HSCs and selecting for proliferation of premalignant pluripotent
stem cells. RHOA mutations occur in TFH cells and NOTCH1 mutations in B cells. Concomitantly, microenvironment interactions, such as communication between
various cells in the germinal center of lymph nodes, including high endothelial venules surrounding dendritic cells, dendritic cells and TFH cells, as well as TFH and
B cells via various cytokines contributes to development of AITL. TFH cells might select for the proliferation and survival of polyclonal verses monoclonal plasma
cells resulting in plasmacytosis associated with AITL.
In this case, it is also possible that the two cancers have arisen independently of one other, or that AITL developed secondary to the chemotherapy treatment of multiple myeloma. Chemotherapy regimens may alter the microenvironment around T cells favoring the proliferation of TFH and subsequent AITL development. However, he received bortezomib which has not been linked to secondary primary cancers. Overall, understanding the relationship between TFH and plasma cells in AITL pathogenesis remains an interesting feature to be pursued to determine if it affects treatment options and overall survival.
Conclusion
This case illustrates a rare presentation of Angio immunoblastic T cell lymphoma developing in a patient who was previously treated for multiple myeloma and relapsed with a new AITL diagnosis. This highlights the importance of understanding the clinical relevance of the relationship between TFH and plasma cells to determine if it can be therapeutically targeted in AITL. Furthermore, identifying high risk cytogenetics associated with hematologic malignancies might allow for improved screening and early detection of these cancers.
References
- Lunning MA, Vose JM. Angioimmunoblastic T-cell lymphoma: the manyfaced lymphoma. Blood. 2017; 129: 1095-1102.
- Ahsanuddin AN, Brynes RK, Li S. Peripheral blood polyclonal plasmacytosis mimicking plasma cell leukemia in patients with angioimmunoblastic T-cell lymphoma: report of 3 cases and review of the literature. Int J Clin Exp Pathol. 2011; 4: 416-420.
- Balagué O, Martínez A, Colomo L. Epstein-Barr virus negative clonal plasma cell proliferations and lymphomas in peripheral T-cell lymphomas: a phenomenon with distinctive clinicopathologic features. Am J Surg Pathol. 2007; 31: 1310-1322.
- Huppmann AR, Roullet MR, Raffeld M, Jaffe ES. Angioimmunoblastic T-cell lymphoma partially obscured by an Epstein-Barr virus-negative clonal plasma cell proliferation. J Clin Oncol. 2013; 31: e28-30.
- Nagoshi H, Kuroda J, Kobayashi T. Clinical manifestation of angioimmunoblastic T-cell lymphoma with exuberant plasmacytosis. Int J Hematol. 2013; 98: 366-374.
- Mitsuhashi K, Shiseki M, Ishiyama M. Angioimmunoblastic T-cell lymphoma with marked polyclonal plasmacytosis in peripheral blood and bone marrow mimicking plasma cell leukemia. Rinsho Ketsueki. 2011; 52: 563-569.
- Xu J, Tang Y, Zhao S. Angioimmunoblastic T-cell lymphoma with coexisting plasma cell myeloma: a case report and review of the literature. Tohoku J Exp Med. 2015; 235: 283-288.
- Suárez AE, Artiga MJ, Santonja C. Angioimmunoblastic T-cell lymphoma with a clonal plasma cell proliferation that underwent immunoglobulin isotype switch in the skin, coinciding with cutaneous disease progression. J Cutan Pathol. 2016; 43: 1203-1210.
- Sakai H, Tanaka H, Katsurada T, Yoshida Y, Okamoto E, Ohno H. Angioimmunoblastic T-cell lymphoma initially presenting with replacement of bone marrow and peripheral plasmacytosis. Intern Med. 2007; 46: 419-424.
- Yamane A, Awaya N, Shimizu T, Ikeda Y, Okamoto S. Angioimmunoblastic T-cell lymphoma with polyclonal proliferation of plasma cells in peripheral blood and marrow. Acta Haematol. 2007; 117: 74-77.
- Singh N, Sharma A, Pasricha S. Florid Plasmacytosis in Angioimmunoblastic T Cell Lymphoma: A Diagnostic Conundrum. Indian J Hematol Blood Transfus. 2018; 34: 188-190.
- Mukherjee T, Dutta R, Pramanik S. Aggressive Angioimmunoblastic T Cell Lymphomas (AITL) with Soft Tissue Extranodal Mass Varied Histopathological Patterns with Peripheral Blood, Bone Marrow, and Splenic Involvement and Review of Literature. Indian J Surg Oncol. 2018; 9: 11-14.
- Jang MA, Lee ST, Kim HJ, Kim S, Kim SH. Simultaneous occurrence of angioimmunoblastic T-cell lymphoma and plasma cell leukemia. Ann Lab Med. 2015; 35: 149-151.
- Fujisawa M, Chiba S, Sakata-Yanagimoto M. Recent Progress in the Understanding of Angioimmunoblastic T-cell Lymphoma. J Clin Exp Hematop. 2017; 57: 109-119.
- Cortés JR, Palomero T. The curious origins of angioimmunoblastic T-cell lymphoma. Curr Opin Hematol. 2016; 23: 434-443.