
Case Presentation
Ann Hematol Oncol. 2019; 6(9): 1270.
Primary Histiocytic Sarcoma of Bone with Disseminated Involvement - Initial Presentation Mimicking Multiple Myeloma: The Atypical Face of a Rare Neoplasm
Lage LAPC1*, Albuquerque VJ1, Moraes RDR2, Couto SCF1, Culler HF2, Rocha V1 and Pereira J1
¹Department of Hematology, Hemotherapy and Cell Therapy, School of Medicine, University of São Paulo (FMUSP), UK
²Department of Pathology, School of Medicine, University of São Paulo (FMUSP), UK
*Corresponding author: Lage LAPC, Department of Hematology, Hemotherapy and Cell Therapy, School of Medicine, University of São Paulo (FMUSP), Av. Dr. Enéas Carvalho de Aguiar, 155 - Cerqueira César, São Paulo - SP, Brazil
Received: September 16, 2019; Accepted: November 12, 2019; Published: November 19, 2019
Abstract
Histiocytic Sarcoma (HS) is an uncommon malignancy derived from macrophage/dendritic lineage cells and accounts for less than 1% of hematologic tumors. Due to its rarity, little is known about its clinical-biological behavior and therapeutic recommendations have not been standardized so far. In addition to lymph node enlargement most cases present extranodal involvement, especially in the gastrointestinal tract, skin and soft tissues. Bone involvement is usually described in association with systemic disease and in advanced stages. Here we describe a case of patient with HS with primary bone involvement and multiple osteolytic lesions and bone fractures in the absence of another organs involvement. A male patient presented with an aggressive disease with rapid progression and his disease was unresponsive to standard therapy. Although rare, this kind of HS presentation should fall within the scope of differential diagnosis of other neoplasms with widespread bone involvement such as multiple myeloma and bone metastasis from solid tumors as the prostate, breast and lungs.
Keywords: Histiocytic sarcoma; Cortical bone; Lytic lesions; Multiple myeloma; BRAFV600E
Abbreviations
HS: Histiocytic Sarcoma; CD: Cluster Designation; KMT2D: histone-lysine N-methyltransferase 2D; MAP kinase: Mitogen- Activated Protein Kinase; MAP2K1: Mitogen-Activated Protein Kinase 1; Hb: Hemoglobin; Ht: Hematocrit; MCV: Mean Cell Volume; MCH: Mean Cell Hemoglobin; MCHC: Mean Corpuscular Hemoglobin Concentration; RDW: Red Cell Distribution Width; WBC: White Blood Count; TIBC: Total Iron-Binding Capacity; ESR: Erythroid Sedimentation Rate; CRP: C-Reactive Protein; Ca: Calcium; HIV: Human Immunodeficiency Virus; MRI: Magnetic Resonance Imaging; CT: Computerized Tomography; H&E: Haematoxyline & Eosin staining; TdT: Terminal Deoxynucleotidyl Transferase; 18-FDGPETCT: 18-Fluorodeoxyglucose-Computerized Tomography With Positron Emission; Max: Maximum; SUV: Standardized Up- Take Value; CHOEP: Cyclophosphamide, doxorubicin, Oncovin, Etoposide and Prednisone; G-CSF: Granulocyte Colony Stimulating Factor; OS: Overall Survival; ICE: Ifosfamide, Carboplatin and Etoposide
Introduction
Histiocytic Sarcoma (HS) is an extremely rare neoplasm responsible for less than 1% of all hematologic malignancies [1]. Although its biological behavior resembles that of lymphomas, its cell line is histiocytic and non-lymphoid. Thereafter, the malignant cell of the HS exhibits morphological and immunophenotypic characteristics of the mature histiocyte. In the most recent 2016 revision of the World Health Organization Classification, histiocytic sarcoma was classified inside the macrophage/dendritic tumors along with others histiocytosis and stroma-derived dendritic cell tumors [2,3].
In the immunohistochemical analysis HS malignant cells shown positivity for one or more histiocytic lineage markers including CD68 (KP1, PGM1), CD163 and lysozyme. CD45, CD45RO and HLA-DR are often positive and it is typically negative for Langerhans cell markers and other dendritic cell markers such as CD1a, langherin, CD21 and CD35 as well as for myeloid, B-lymphoid and T-lymphoid markers. S-100 protein may be positive but usually it expression is weak or focal and Ki67 has a variable expression [4].
Gene expression studies have shown that a subset of HS has clonal rearrangement of the immunoglobulin gene, especially in cases associated with low-grade B-cell lymphomas [5]. BRAF V600E mutation has been also described in some cases. In a recent study 5 of 8 cases of HS presented the BRAF V600E mutation, making this information a promising option for target therapy [6]. In addition, recurrent mutations involving the KMT2D gene and MAP kinase pathway genes including KRAS, NRAS and MAP2K1 have been described [7,8]. Despite the progress made in the molecular field, its etiology still remains unknown.
HS is usually aggressive and present as advanced stages III and IV and with poor response to therapy, although exceptions have been reported. In fact, most patients die from disease progression [9]. Lymphadenopathy is common and most cases present extralymphonodal involvement, most commonly in the gastrointestinal tract, skin, superficial and deep soft tissues, lung and nasal cavity [10]. Few reports have shown neoplasia arising in the central nervous system, endocrine tissues and bone marrow [11,12]. Rarely, patients shown systemic disease with multiple disease sites referred to as malignant histiocytosis.
Secondary bone involvement is described in systemic disease as well as in advanced stages, but primary bone disease was not described in the literature yet. Here, we report the case of a 32-yearold man with primary bone HS presenting with disseminated bone involvement characterized by multiple lytic lesions and fractures mimicking multiple myeloma. This is the first case of primary bone HS reported to date, an extremely rare and aggressive form of this disease.
Case Report
A previously healthy 32-year-old Brazilian man has a 4-month history of weight loss of 33 kg, night sweats, intermittent fever, fatigue, intolerance to usual efforts, and solid tumors onset in the trunk and scalp. He evolved with significant bone pain in the scapula, sternum, ribs and lumbosacral spine. The patient denied perception of lymph node enlargement, pruritus, as well as a history of recurrent infections and hemorrhagic manifestations.
He was admitted in our emergency department complaining loss of strength and sensitivity in both lower limbs, associated with urinary retention and fecal incontinence with started 1 week ago. Physical and neurological examination revealed deep skin and mucous pallor, flaccid crural paraplegia and hardened tumors on the back, scalp, sternum and left costal gradient. There were no superficial lymphadenopathy or hepatosplenomegaly.
Initial laboratory tests showed: Hb 63 g/L, Ht 19%, MCV 82 fl, MCH 29 pg, MCHC 33%, RDW 15.4%, WBC 14.09 x 109/L (73.1% neutrophils, 4.0% eosinophils, 0.1% basophils, 15.7% lymphocytes and 7.1% monocytes) and platelet count of 722 x 109/L. Reticulocyte count 1.15% (30,000 reticulocytes/mm3) and inflammation-consistent iron profile (serum iron: 34 μg/dL, iron saturation 17%, ferritin 6,795 ng/mL, transferrin 158 mg/dL and TIBC = 202 μg/dL). Renal, hepatic and thyroid function within normal range but with increased lactic dehydrogenase (492 U/L) and significant increase of inflammatory tests (ESR = 140 mm/h and CRP = 252.3 mg/L). He also presented with hypercalcemia (ionic Ca++ 5.44 mg/dl) and polyclonal hypergammaglobulinemia. HIV, B and C hepatitis serologies were negative.
Upon clinical suspicion of complete spinal cord compression syndrome, the patient underwent thoracic and lumbosacral Magnetic Resonance Imaging (MRI), which showed multiple osteolytic lesions diffusely affecting the bone and vertebral bodies, some with cortical rupture and soft tissue component, as well as vertebral fractures compromising T4, T6 and T10 with evidence of compressive myelopathy. Chest, abdomen and pelvis CT scans revealed osteolytic lesions in pelvic bones and a large mass on the left tenth rib with a soft tissue component measuring 9.2 x 6.0 x 7.8 cm and a soft tissue component mass involving the iliac and sacrum bone with evidence of foraminal invasion, measuring 10.9 x 9.0 x 5.1 cm.
Bone biopsy showed undifferentiated malignant proliferation of mononuclear cells with high histological grade and necrosis (H&E - Figure 1A). Cell proliferation presented plasmacytoid/ cuboidal morphological differentiation in a solid and linear discoid arrangement, with some large bi or multinucleated atypical cells, with scarce cytoplasm and minimal inflammatory background. The neoplastic cells were immunohistochemically positive for vimentin, lysozyme, diffuse CD68, CD45, CD43, CD99 and ki67 in 50% of the cells and negative for myeloid, lymphoid B and T-associated markers, cytokeratin, CD15, CD30, CD34, CD56, CD1a, CD138, S-100, TdT, desmina and Melan-A (Figure 1 - B, C and D), allowing the HS diagnosis.
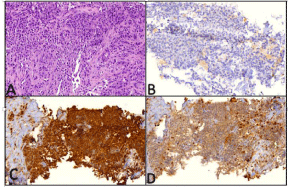
Figure 1: (A) Undifferentiated malignant neoplasm infiltrating fibro-connective
tissue (H&E; 10x optic microscope). (B) CD1a negative immunohistochemical
labeling. (C) Diffuse positive staining for CD68, dense granular cytoplasmic
pattern. (D) Positive staining for lysozyme, cytoplasmic pattern.
Additional investigation was performed, including bone marrow aspirate and biopsy, which demonstrated normocellular bone marrow, with norm maturation of the 3 medullary sectors and absence of anomalous or neoplastic cells. Immunophenotyping of bone marrow by flow cytometry showed no changes and cytogenetic study showed no metaphases. Screening for BRAF V600E mutation was negative in peripheral blood and tumor bone specimen in paraffin-embedded material, using second generation sequencing by the Tumor Hotspot MASTR Plus/Agilent assay (BRAF gene, codon 600: wild-type). Immunohistochemical staining of the mutated BRAF protein was also negative in the neoplastic tissue studied.
Staging was performed with 18-FDG-PETCT showing multiple osteolytic lesions with marked glycolytic hypermetabolism, some with cortical rupture and extra-bone soft tissue component. Some lesions showed central hypometabolic areas corresponding to necrosis in particular in the lateral segment of the 10th right costal arch measuring 4.0 x 2.0 cm (standardized uptake value/Max SUV of 17.5), 11th and 12th left costal arches measuring 9.2 x 6.0 x 7.8 cm (maxSUV: 19.2), distal portion of the sternum and xiphoid process measuring 6.2 x 4.3 x 4.6 cm (maxSUV: 21.1), iliac and sacrum associated with an important extra osseous soft tissue component with signs of invasion of the right sacral neural foramen measuring 10.9 x 9.0 x 5.1 cm (maxSUV: 11.9). Other predominantly hypermetabolic lesions were observed in the axial and appendicular skeleton, scalp, scapula, clavicles, humerus, manubrium, body of the sternum, costal arches, spine (cervical, thoracic and lumbosacral), ilium, pubis, bilateral acetabulum and femur (maxSUV: 22.5 - left ilium) – (Figure 2A).
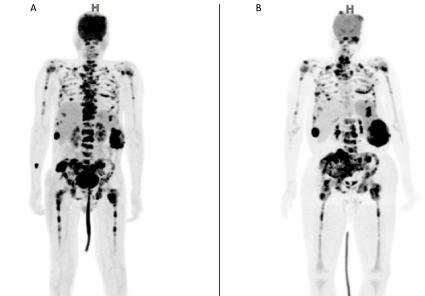
Figure 2: 18-FDG-PETCT before (A) and after (B) two cycles of CHOEP
multidrug therapy. High FDG uptake in multiple bones and extraosseous soft
tissue components. In (B) there is an increase in 18-FDG uptake and tumor
size, consistent with primary refractory disease.
After diagnosis of spinal cord compression syndrome and diffuse bone HS the patient received local radiotherapy from T5 to T11 (3.000cGy), associated with parenteral esteroids without any neurological improvement. Subsequently, he received CHOEP-21 protocol chemotherapy (cyclophosphamide 750 mg/m2 IV D1, doxorubicin 50 mg/m2 IV D1, etoposide 100 mg/m2 IV D1 to D3, vincristine 1.4 mg/m2 [Max 2.0 mg] IV D1 and prednisone 100 mg/ day PO D1 to D5). After two courses of chemotherapy he presented progression of bone tumors and remained felling painful. Then, a new 18-FDG PETCT was performed which demonstrated increased size and degree of glycolytic metabolism in bone and soft tissue components compatible with refractory disease (Figure 2B). Salvage treatment consisted of cladribine and high doses of cytarabine and G-CSF. After 15 days, the patient developed refractory septic shock due to multiresistant Pseudomonas aeruginosa dying after 3 months of hospitalization.
Discussion
HS is an extremely rare neoplasm, with a limited number of case series reported in the medical literature so far. Thus, little is known about its clinical presentation, cell biology and therapeutic management. Most reported cases occur in adults however the disease can be diagnosed in all age groups from childhood to elderly. The case series shown a median age at diagnosis ranging from 46 to 63 years old (18 - 96 years). Data from the US Surveillance Epidemiology registry show a predilection for males in a total of 159 cases of HS, 99 men and 60 women [13].
The diagnosis of HS is based on the histopathological and immunohistochemical findings in association with clinical symptoms and laboratory data. The usefulness of flow cytometry in identifying this neoplasm is unclear, but can sometimes be helpful in supplementing the diagnosis. Bone marrow evaluation is necessary in all suspected cases since if there is infiltration by myeloid blasts with monocytic differentiation, the diagnosis to be established will be with granulocytic sarcoma with monocytic differentiation, the main differential diagnosis of HS [1].
In our case, the histopathological and immunohistochemical findings of a thick-needle biopsied tumor specimen supported the diagnosis of HS. Neoplastic cells were positive for more than one histiocytic marker, including CD68 and lysozyme, and were typically negative for markers of myeloid lineage, lymphoids B and T, follicular dendritic cells, and Langerhans cells, thus indicating definitive diagnosis of HS. Bone marrow aspirate and biopsy showed no involvement by disease ruling out the diagnosis of granulocytic sarcoma with monocytoid differentiation associated with monocytic acute leukemia.
In a recent study published in 2018, Brodwater et al. described 23 cases of HS, isolated bone involvement was not described in any of these patients [9]. In the largest case series published to date comprising 159 cases of HS from several North American centers, primary bone involvement has not been described by the authors either. The most frequent sites involved by the disease were connective tissue and skin (35.8%), lymph nodes (17%), respiratory tract (8.2%), central nervous system (6.9%), gastrointestinal tract (7.5%) and hematopoietic system/spleen (9.4%) [13]. Our report has been proposed a new form of HS not described in the literature so far, with multiple osteolytic lesions, extraosseous soft tissue component and vertebral fractures, mimicking presentation of advanced multiple myeloma. No other organic involvement was evidenced that could justify bone lesions as secondary involvement.
Many reports have described the appearance of HS following or simultaneously with other neoplasias including solid tumors (breast, lung, colon) and hematologic malignancies, such as follicular lymphoma, myelodysplasia and acute lymphoblastic leukemia [14,15]. Thus HS can be classified as primary or secondary. In the case series of Brodwater et al., (N = 23) secondary HS presented with more aggressive behavior than de novo HS (11.8 months OS compared to 70 months, with a median difference of 58.2 months, p = 0.001) [9]. Although our case was characterized as de novo HS, its poor prognosis (OS 4 months) and marked clinical aggressiveness leads us to believe that disseminated primary bone involvement is a factor related to poor prognosis, similar to that observed in secondary HS cases.
Clinical presentation of this malignancy varies from localized nodal involvement to disseminated and rapidly progressive extranodal disease. Patients may have solitary mass, fatigue, and systemic symptoms such as fever, weight loss, and night sweats. Skin manifestations can range from a seemingly benign rash to numerous skin tumors. Hepatosplenomegaly and pancytopenia may occur. Intestinal lesions often present with obstruction or perforation. Spinal cord compression syndrome has not been described so far as being a HS-related oncological complication which makes our case quite peculiar.
Due to its rarity, no standard treatment has been established for HS. Therapy data are limited and result from case reports or small series of patients. In addition, the natural history of HS is largely unknown and usually variable. The choice between therapeutic options is primarily based on the extent of the disease, patient performance status and medical team experience. Apparently, for localized disease the best choice of therapy is surgical resection, associated or not with radiotherapy. On the other hand, treatment usually used for advanced disease consists of systemic chemotherapy, using CHOP or ICE as the main options, according to with the patient’s performance status. Such treatments are based on a limited number of case reports that have been successful with these approaches [16,17].
The patient in this case had advanced and aggressive systemic disease, so we opted for standard chemotherapy with CHOP more etoposide (CHOEP) regimen. More aggressive chemotherapy, such as ICE regimen, was not applied due to our patient’s compromised performance status, which led us to believe that he would not tolerate intensified approaches. Despite the therapy employed, after two cycles of chemotherapy no response was achieved, thus characterizing refractory disease.
Patients with HS who relapse or fail to achieve response after first-line chemotherapy may require additional therapies such as second-line chemotherapy, including high-dose cytosine arabinoside and purine analogues followed by consolidation with autologous or allogeneic hematopoietic stem cell transplantation [16,18,19]. Our patient experienced a cycle of high-dose cladribine and cytarabine chemotherapy but evolved soon after with death due to infectious complications inherent to treatment.
Thalidomide therapy, alemtuzumab (anti-CD52 monoclonal antibody) and other therapies including vemurafenib (BRAF inhibitor), imatinib, sorafenib and bevacizumab have also been employed in some cases; however, treatment response is usually poor [18].
Histiocytic diseases presenting the BRAF V600E mutation appear to have a good response to BRAF inhibitors such as vemurafenib, although the activity of these drugs in HS is not yet well documented. At present, vemurafenib and dabrafenib cannot be recommended as first-line therapy in HS. However, patients with BRAFV600E mutation HS who have relapsed/refractory to previous systemic therapy can reasonably be treated with vemurafenib or dabrafenib [20]. In our case the molecular investigation for BRAFV600E mutation was negative, making this therapeutic option inappropriate. Autologous or allogeneic hematopoietic stem cell transplantation should be considered for patients with relapsed/ refractory disease who achieve remission after salvage therapy.
In conclusion, to our knowledge, this is the first case of primary HS with disseminated bone involvement. The existence of this entity can be confirmed by the typical findings of biopsy and expression of histiocytic markers in the immunohistochemical study, as well as by the clinical picture and imaging exams that did not show evidence of other organs involved in the neoplasia. This finding allows that primary bone HS, although rare, be incorporated in the scope of the differential diagnosis of diseases with disseminated osteolytic lesions, such as multiple myeloma and bone metastases of solid tumors.
References
- Swerdlow S, Campo E, Harris NL, Jaffe ES, Pileri S, Stein H, et al. WHO classification of tumors of haematopoietic and lymphoid tissues. IARC Press, Lyon. 2017.
- Wang E, Hutchinson CB, Huang Q. Histiocytic sarcoma arising in indolent small B-cell lymphoma: report of two cases with molecular/genetic evidence suggestive of a “transdifferentiation” during the clonal evolution. Leuk Lymphoma. 2010: 51: 802-812.
- Gounder M, Desai V, Kuk D. Impact of surgery, radiation and systemic therapy on outcomes of patients with dendritic cell and histiocytic sarcomas. Eur J Cancer. 2015; 51: 2413-2422.
- Pileri SA, Grogan TM, Harris NL. Tumors of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology. 2002; 41: 1-29.
- Takahashi E, Nakamura S. Histiocytic sarcoma: na update literature review based on the 2008 WHO classification. J Clin Exp Hematop. 2013; 53: 1-8.
- Go H, Jeon YK, Huh J. Frequent detection of BRAF (V600E) mutations in histiocytic and dendritic cell neoplasms. Histopathology. 2014; 65: 261-272.
- Hung YP, Lovitch SB, Qian X. Histiocytic sarcoma: new insights into FNA cytomorphology and molecular characteristics. Cancer Cytopathol. 2017; 125: 604-614.
- Shanmugam V, Sholl LM, Fletcher CD. RAS/MAPK pathway activation defines a commom molecular subtype of histiocytic sarcoma. Mod Pathol. 2018; 31: 551.
- Broadwater DR, Conant JL, Czuchlewski DR. Clinicopathologic features and clinical outcome differences in de novo versus secondary hystiocytic sarcomas: a multi-institutional experience and review of the literature. Clinical Lymph, Myeloma & Leuk. 2018; 18: 427-435.
- Hung YP, Qian X. Histiocytic sarcoma. Arch Pathol & Lab Med. 2018; 1: 1-5.
- Foster M, Kamaly-Asl I, Stivaros S. Primary cerebral histiocytic sarcoma in childhood: a case report of protracted survival and review of literature. Childs Nerv Syst. 2015; 31: 2363-2368.
- Zanelli M, Ragazzi M, Marchetti G. Primary histiocytic sarcoma presenting as diffuse leptomengial disease: case description and review of the literature. Neuropathology. 2017; 37: 517-525.
- Kommalapati A, Tella SH, Durkin M. Histiocytic sarcoma: a population-based analysis in incidence, demographic disparities, and long-term outcomes. Blood. 2018; 131: 265-268.
- Geyer JT, Yigit M, Miyaguchi A. Histiocytic sarcoma following B-lymphoblastic leukemia/lymphoma. Am J Clin Pathol. 2019.
- Farris M, Hughes RT, Lamar Z. Histiocytic sarcoma associated with follicular lymphoma: evidence for dramatic response with rituximab and bendamustine alone and a review of literature. Clin Lymphoma, Myeloma Leuk. 2019; 19: e1-e8.
- Tsujimura H, Myiaki T, Yamada S. Successful treatment of histiocytic sarcoma with induction chemotherapy consisting of dose-escalated CHOP plus etoposide and up-front consolidation auto-transplantation. Int H Hematol. 2014; 100: 507-510.
- Chen X, Zhang L, Wang J. Complete response after chemotherapy and radiotherapy of a tonsilar histiocytic sarcoma with regional lymph node involvement: a case report and review of literature. Int J Clin Exp Med. 2015; 8: 16808-16812.
- Cruz-Chacon A, Mathews J, Ayala E. Transplantation in rare lymphoproliferative and histiocytic disorders. Cancer Control. 2014; 21: 335- 342.
- Tomlim J, Orosco RK, Boles S. Successful treatment of multifocal histiocytic sarcoma occurring after renal transplantation with cladribine, high-dose cytarabine, G-CSF, and mitoxantrone (CLAG-M) followed by allogeneic hematopoietic stem cell transplantation. Case Report Hematol. 2015; 20: 72- 82.
- Kordes M, Röring M, Heining C. Cooperation of BRAF (F595L) and mutant HRAS in histiocytic sarcoma provides new insights into oncogenic BRAF signaling. Leukemia. 2016; 30: 937-946.