
Perspective
Ann Hematol Oncol. 2020; 7(8): 1318.
Myelodysplastic Syndromes: A Diagnostic and Therapeutic Conundrum
Islam A*
Division of Hematology/Oncology, Department of Medicine, Buffalo General Hospital, Buffalo, New York
*Corresponding author: Anwarul Islam M.D., Division of Hematology/Oncology, Department of Medicine, Buffalo General Hospital, Room E 318, Buffalo, New York 14203, New York
Received: October 30, 2020; Accepted: December 03, 2020; Published: December 10, 2020
Perspective
Hematological malignancies are a diverse group of neoplasms that affect the blood, bone marrow and lymphatic systems [1], and Myelodysplastic Syndromes (MDS) are considered as belonging to this group [2,3]. Although it is debatable whether MDS are neoplasms of the hematopoietic system or a reflection of an underlying disorder, it is recognized that some but not all do develop into acute leukemia and in that sense they can be considered as “pre-leukemia”, the term that was replaced for MDS by the FAB classification [4].
Laboratory diagnosis of MDS is almost exclusively based on the morphological features of hematopoietic cells observed in smears of peripheral blood and bone marrow, and this depends solely on the presence of dysplastic changes observed in the granuloid, erythroid, and megakaryocytic cells in the absence of iron, vitamin B12 or folic acid deficiency. Hematoxylin and Eosin (H&E) stained sections of paraffin-embedded bone marrow trephine biopsies are usually insufficient to clearly detect such characteristic morphologic changes, though they can perhaps demonstrate particular abnormalities in megakaryocytes, such as micromegakaryocytes, large mononuclear forms, bi or multinucleated forms and the localization of clusters of immature myeloid precursor cells (blast cells) in some areas of the section. In the original and later classifications of MDS, evidence discovered in the sections of bone marrow biopsy specimens was not taken into account, although such evidence might have provided valuable diagnostic and prognostic information [5,6]. In recent years attempts have been made to diagnose MDS not only based on the morphology alone but also including cytogenetic and molecular markers [7]. However, under most circumstances and in daily practice the diagnosis of MDS continues be exclusively based on the dysplastic morphology of hematopoietic cells observed in the smears of peripheral blood and bone marrow.
Common morphologic findings in MDS are summarized table 1. For the neutrophils, hypogranularity and hypolobation may be observed in blood smears, while giant metamyelocytes and bizarre nuclear forms can occur in the bone marrow (Figure 1). The erythroid abnormalities in the peripheral blood smear include anisocytosis, poikilocytosis, ovalocytes, macro-ovalocytes and dimorphic picture (Figure 2). The bone marrow aspirate smear may include members of the erythroid series displaying megaloblastoid features, altered nuclear-cytoplasmic ratios (nuclear cytoplasmic asynchrony), nuclear budding, bilobed nuclei, nuclei with an irregular outline, cytoplasmic vacuoles, inter-nuclear and inter-cytoplasmic bridging, (Figures 3 & 4) and sideroblasts including ring sideroblasts (Figure 5). The morphological abnormalities of the megakaryocytic series observed in the peripheral blood include giant platelets (Figure 2) while bone marrow aspirates and bone marrow biopsy sections may include micromegakaryocytes, excessive multinuclearity or hypolobulated megakaryocytes (Figure 6).
![]()
Dyserythropoiesis
Dysgranulopoiesis
Dysmegakaryopoiesis
Peripheral Blood
Anisocytosis Poikilocytosis Basophilic Stippling Ovalocytes Macro-Ovalocytes Dimorphic Picture
Nuclear Hypolobulation (Pseudo-Pelger-Huet Anomaly) Hyperlobulation Hypogranulation
Platelet Anisocytosis Giant Platelets
Bone Marrow
Eythroblasts With Excess Cytoplasm Nuclear Budding Irregular Nuclear Outline Inter Nuclear Bridging Intercytoplasmic Bridging Bi and Multinuclearity Nuclear Lobulation Megaloblastoid Changes Cytoplasmic Vacuolization Cytoplasmic Inclutions Nuclear Cytoplasmic Asynchrony Karyorrhexis Multinucleated Megaloblastoid Erythroid Precursors Ring Sideroblasts
Nuclear Hypolobulation (Pseudo-Pelger-Huet Anomaly) Hyperlobulation Hypogranulation Bizarre Nuclear Shape Giant Metamyelocytes
Micromegakaryocytes Single Small Nucleus Nuclear Hypolobulation Small Binucleated Form Multinuclear Forms
Table 1: Common Morphologic Changes Observed in Erythroid, Granulod and Megakaryocytic Cells.
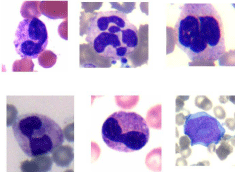
Figure 1: Neutrophilic dysgranulopoiesis observed in the blood and bone
marrow. Note the pseudo Pelger-Huet anomaly, bilobed and ‘spectacle’
forms, and bizarre nuclei along with one blast cell (lower right corner).
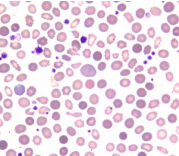
Figure 2: Dyserythropoiesis: peripheral blood smear showing dimorphic
blood picture along with anisocytosis, poikilocytosis and presence of
ovalocytes, macro-ovalocytes (one in the center).
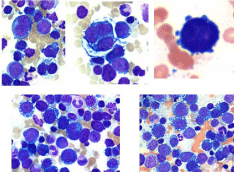
Figure 3: Dyserythropoiesis bone marrow aspirate smear showing mono and
multinucleated megaloblastoid erythroid precursors, cytoplasmic blebs and
vacuolation.
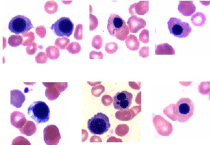
Figure 4: Dyserythropoiesis: bone marrow aspirate smear showing intercytoplasmic
bridging, altered nuclear: cytoplasmic ratios, nuclear cytoplasmic
maturation asynchrony, cytoplasmic vacuolation, nuclear budding.
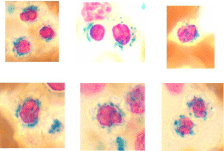
Figure 5: Photomicrographs showing sideroblasts and ring sideroblasts in
the bone marrow aspirate smear.
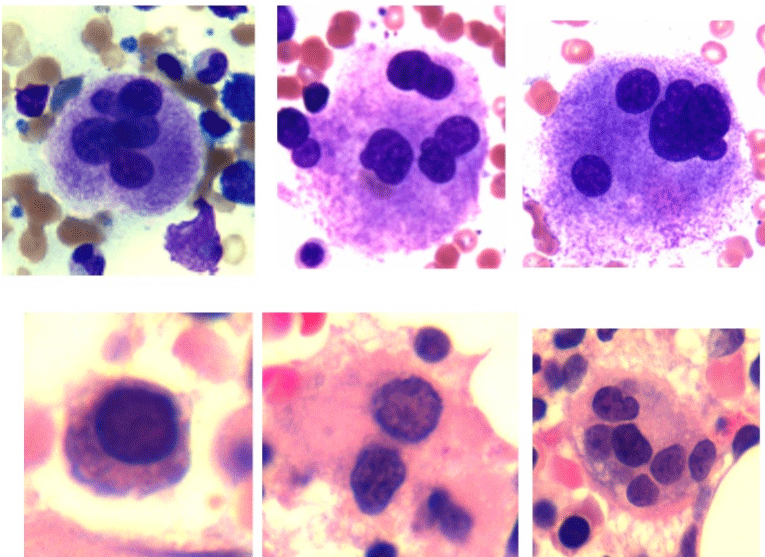
Figure 6: Dysmegakaryopoiesis: multi nuclear megakaryocytes, top rowbone
marrow aspirate smear. Mono, bi and multi-nucleated megakaryocytes,
bottom row- bone marrow biopsy section.
A diagnosis of MDS is typically made by practicing hematologists in Europe and mostly by hematopathologists in the United States by examining the smears of peripheral blood and bone marrow aspirates. Evaluation of sections of bone marrow trephine biopsy specimens does not yet play a pivotal role in its diagnosis. Unlike acute myeloid leukemia which might present a marrow that is heavily infiltrated with myeloblasts and is readily diagnosable, MDS is likely to display a varied picture involving one or more of the granular, erythroid and megakaryocytic populations. This profile does not lend itself to a specific, exclusive categorization. This can result in multiple varied assessments from different independent hematologists or hematopathologists. Inter-observer variation in the diagnosis of MDS [8] and difficulty in distinguishing genuine MDS from mimics [9] is not uncommon. For example, a group of hematologists or hematopathologists can describe one bone marrow independently in many different ways, just as six blind people described an elephant in many different ways [10]. In addition, bone marrow is not always involved uniformly in pathological processes, and morphological discordance is well known [11,12]. This is particularly true for patients with myelodysplastic syndromes. In bone marrow biopsy sections from patients with MDS one could see a non-uniform involvement of the marrow with different cellular elements including localized clusters of blast cells in different areas of the same section. The latter constellation has been termed ‘Abnormal Localization of Immature Precursors (ALIP)’ [13] (Figure 7).
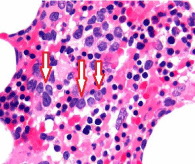
Figure 7: Abnormal Localization of Immature Precursors (ALIP) (arrows)
observed in the bone marrow biopsy section from a patient with MDS (H&E).
During bone marrow aspiration, if the tip of the aspirating needle encounters one of these ‘abnormal localization of immature precursors,’ (Figure 8) the aspirate may contain 15-20% or more blast cells and the patient might then be misleadingly diagnosed as having high risk MDS or even de-novo acute leukemia. On the other hand, during aspiration if the tip of the aspirating needle misses these areas of focal collection of blast cells (Figure 8) and strikes an area containing clusters of erythroid precursors as shown in the picture (or where erythroid precursors predominate) such bone marrow aspirates will definitely have few blast cells and the patient would be considered having a low risk of MDS. Alternatively in this situation the case could be misleadingly diagnosed as erythroleukemia (FAB M6).
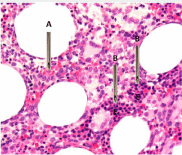
Figure 8: Same biopsy section as in figure 7 but lower magnification
demonstrating. A- the tip of aspirating needle has encountered an area
where clusters of immature myeloid precursors (blast cells) are present. They
can be easily identified by the presence of nucleoli. B- demonstrates where
the tip of the aspirating needle has encountered an area where blast cells are
absent but predominates with erythroid precursors (E).
MDS is primarily a disease of the elderly and most patients are at 65 or over. The disease is characterized by ineffective hematopoiesis and bone marrow failure. Deficiencies in hematopoiesis usually result in peripheral blood cytopenias, of which anemia is the most common. However, neutropenia and thrombocytopenia, either singly or in combination, may dominate the blood picture. The bone marrow usually is hypocellular but sometimes it may be normocellular or hypercellular which may lead to its interpretation or assessment as a myeloproliferative disorder and misleadingly diagnosed as Myelodysplastic/Myeloproliferative Neoplasm (MDS/MPN) [14]. The term MDS/MPN is another diagnostic conundrum that the international working group has led the practicing hematologist into [14]. There is no justification for incorporating myeloproliferative disorders into the category of myelodysplastic syndromes. All patients with myeloproliferative disorder show some degree of myelodysplastic changes and it is perhaps scientifically incorrect and diagnostically ambiguous to combine these two possibly separate diseases under one umbrella.
We have diligently studied well stained bone marrow aspirate and plastic embedded bone marrow biopsy sections from normal adults as well as from patients with various hematological disorders [15]. Normal marrows (aspirates and trephine biopsies) were obtained from normal adults who volunteered to donate marrow for transplantation. Patients with AML, for example were studied at diagnosis, following chemotherapy, during hematopoietic regeneration following chemotherapy and at complete remission [16-18]. Patients with Chronic Granulocytic Leukemia (CGL) were studied in the chronic phase, during blast cell transformation, and following autographing and during hematopoietic regeneration [19- 21]. Bone marrow aspirates and biopsies were also obtained from patients with aplastic anemia, chronic lymphocytic leukemia, non- Hodgkin’s lymphoma and multiple myeloma.
We have observed that some degree of myelodysplastic alteration either in the erythroid, granuloid or megakaryocytic series can be observed in most of these disorders. Even in normal marrows, some dysplastic changes in the erythroid, granuloid or megakaryocytic cells can be observed but they are few and far between. In the original FAB classification [4] it was reported that “in some patients the mere presence of these abnormalities is adequate for their inclusion in the MDS”. This premise requires farther delineation because if this were to be accepted then many of the newly diagnosed patients with AMLs, multiple myelomas, Hodgkin’s lymphomas and other marrow-perturbing neoplastic conditions (where such morphologic abnormalities are not uncommon) would need to be included in MDS.
Cytogenetic abnormalities are present in many of the MDS cases [22] and are believed to have both diagnostic and prognostic significance. Prognostic groups have been proposed on the basis of cytogenetics in the revised international score (IPSS-R) scheme, which include 5 different subgroups including 20 different alterations [23]. The most common cytogenetic abnormalities encountered in MDS are del(5q) [5q-], -7, and -8. Cytogenetic studies are performed on spontaneously dividing cells that are typically cultured for short periods (24-72h). MDS with hypocellular bone marrow may not provide a sufficient number of such spontaneously dividing cells and as a result cytogenetic studies in these cases may not be accurate. Karyotypic changes can also be found in many if not most hematological malignant conditions, and some are non-specific irrespective of the disease in which they are found. Although there is some evidence from both cytogenetic and molecular studies [24] that they have an impact on response and outcome but these findings need to be included in a newly devised, organized classification of MDS to be clinically useful and universally recognized. But until an accurate reproducible diagnostic classification (involving cytology, histology, flow-cytometry, cytogenetics and molecular studies) is established we may not know the actual relevance of these studies with regards to such broad array (enumerated below) of current diagnostic classifications, which are solely based on the morphology of hematopoietic cells observed in the smears of peripheral blood and bone marrow.
In patients with a hypocellular bone marrow it can be difficult to distinguish between severe aplastic anemia and MDS due to the paucity of cells in the aspirate smear and core biopsy to assess dysplastic changes. It has been suggested that in such situations, the presence of abnormal megakaryocytes and MDS-associated cytogenetic changes [25] may favor a diagnosis of MDS. But too often there are hardly any cells on which to carry out appropriate cytogenetic or molecular studies or look for specific dysplastic changes in hematopoietic cells.
Another important diagnostic challenge is to distinguish MDS with bone marrow fibrosis from primary myelofibrosis [25]. Here, the absence of Janus Kinase 2 (Jak2 V617F) mutations and the presence of MDS- associated cytogenetic changes and megakaryocytic dysplasia may be helpful factors in distinguishing MDS from primary myelofibrosis. However, in large majority of cases with essential thrombocythaemia, an excessive proliferation of megakaryocytes occurs; such megakaryocytes are quite often dysplastic and associated with marrow fibrosis. These cases should be diagnosed as a myeloproliferative disorder and not MDS despite the presence of dysplastic megakaryocytes and the absence of Jak2 mutation.
Chronic Myelomonocytic Leukemia (CMML) is believed to be a myeloid neoplasm characterized by an excessive production and accumulation of monocytoid cells in the peripheral blood and bone marrow. This is commonly associated with dysplasia in one or more hematopoietic cell lineages and an increased risk of transformation into acute myeloid leukemia [26,27]. CMML was originally classified as MDS in the FAB classification [4]. However, it has been correctly removed from the current WHO classification. CMML should never have been included in the classification of MDS in the first place. CMML truly belongs to chronic myeloproliferative disorders and the condition is very similar to Chronic Granulocytic Leukemia (CGL), but unlike CGL, where mature granulocytic precursor cells predominate in the bone marrow and peripheral blood, in CMML it is the monocytes and pro-monocytes that predominate in these two sites (Figure 9). Substantial dysplasia in one or more hematopoietic cell lineages are not uncommon in CGL and transformation of CGL to AML known as blast cell crisis is common. The only difference in these two disease entities is perhaps the absence Philadelphia chromosome and its related BCR-ABL1 fusion gene in CMML.
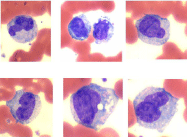
Figure 9: Myelo-Monocytic Leukemia (MMOL): Peripheral blood and bone
marrow aspirate smear showing monocytes and promonocytes.
The presence or absence of ring sideroblasts plays a prominent role in the diagnosis and classification of MDS in the FAB classification [4]. Sideroblasts are nucleated erythroid cells whose cytoplasm contains siderotic (iron positive) granules that are usually well dispersed in the cytoplasm but may also be localized adjacent to the nucleus. Ring sideroblasts display their granules in a perinuclear distribution surrounding the entire nucleus or at least one third of the nucleus. Sideroblasts are found in diverse circumstances, including megaloblastic anemia [28]. The term sideroblastic anemia has been applied to or viewed as a complication of the anemia of severe alcoholism [29], B-6 deficiency [30] and lead poisoning [31]. It is debatable whether the presence of sideroblasts or ring sideroblasts should be afforded any such prominence in the diagnosis and classification of MDS.
MDS is believed to be a group of heterogenous disorders in which the understanding of their pathogenesis is still incomplete. Recent advances have shown that immune dysregulation may contribute to the disease pathobiology in selected individuals [25]. An analysis of the broad array of classifications of MDS [ (i) MDS with single lineage dysplasia -MDS-SLD, (ii) MDS with ring sideroblasts - MDS-RS, (iii) MDS with multilineage dysplasia - MDS-MLD, (iv) MDS with excess blasts-1 -MDS-EB-1, (v) MDS with excess blasts-2 - MDS-EB-2, (vi) MDS unclassifiable - MDS-U, (vii) MDS with isolated del(5q), (viii) refractory cytopenia of childhood, (ix) MDS with excess blasts in transformation –MDS-EB-T] [32,33], indeed leads to the conclusion that a consensus for an accurate, specific, knowledgeable diagnosis of MDS has yet to be met. This might be the reason why the prognosis and clinical course of MDS varies so much among patients. Some transform into leukemia or die from bone marrow failure shortly after diagnosis and others survive for years without major clinical problems [6]. Perhaps the best interim resolution of this terminological quandary is to revert to the prior older classification of “pre-leukemia” and manage these conditions individually, whether they are anemic, neutropenic or thrombocytopenic or of mixed deficiencies. They should not be considered as hemopoietic malignant disorders such as acute leukemia and ought to be treated with chemotherapy. Some of these patients can be watched and observed for a long period of time with regular clinic visits and monitoring only with the CBC with differentials. For patients who require medical treatment, growth factors, such as epoietin and GCF along with a low dose steroid can be effective for those with anemia, neutropenia, or both [34,35]. Even patients with an excess of blast cells in the marrow also respond to above therapy quite well (author’s personal observation). Patients with anemia in whom erythropoietic-stimulating agents have not been effective may benefit from Luspatercept (a recombinant fusion protein that binds transforming growth factor ß superfamily ligands to reduce SMAD2 and SMAD3 signaling) [36]. Immunomodulatory drugs such as revlimid may be useful in some cases [37]. Chemotherapy, at least as an initial treatment may be avoided. A controlled and comparative study of MDS patients with growth factors (G-CSF and erythropoietin), immunomodulatory drugs (revlimid) or chemotherapy “5-azacytidine” would be welcome. But until we are able to formulate an accurate diagnostic criteria beyond morphological grounds alone (which is subject to interpretation) and overcome the inter-observer variation, we are stuck and we will be comparing apples and oranges to formulate an accurate diagnosis. As a result, therapeutic dilemmas will persist. Until we find an adequate solution to this conundrum a wise approach would be to watch and observe and treat these patients only when necessary, either with growth factors or immunomodulatory drugs in combination with supportive therapy such as blood and platelet transfusions. Some of these patients do run very low platelet counts (around 20,000) and fortunately they do not bruise or bleed easily. In such circumstances an intelligent approach would be to leave them alone and not to become overly enthusiastic and aggressively treat the thrombocytopenia.
References
- Snowden JA, O’Connell S, Hawkins J. Haematological cancers: improving outcomes. A summary of updated NICE service guidance in relation to Specialist Integrated Haematological Malignancy Diagnostic Services (SIHMDS). J Clin Pathol. 2017; 70: 461-468.
- Ridgeway JA, Tinsley S, Kurtin SA. Practical Guide to Bone Marrow Sampling for Suspected Myelodysplastic Syndromes. J Adv Pract Oncol. 2017; 8: 29- 39.
- Gangat N, Patnaik MM, Tefferi A. Myelodysplastic Syndromes: Contemporary review and how to treat. 2016; 91: 76-81.
- Bennett JM, Catovsky D, Daniel MT. The French- American-British (FAB) Co-operative group. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982; 51: 189-199.
- Horny HP, Sotlar K, Valent P. Diagnostic value of histology and immunohistochemistry in myelodydplastic syndromes. Leu Res. 2007; 31: 1609-1616.
- Valent P, Horny HP, Bennett JM. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: Consensus statements and report from working conference. Leukemia Research. 2007; 31: 727-736.
- Valent P, Orazi A, Steensma DP. Proposed minimal diagnostic criteria for Myelodysplastic Syndromes (MDS) and potential pre-MDS conditions. Oncotarget. 2017; 5: 73483-73500.
- Font P, Loscertales J, Benavente C. Inter-observer variance with the diagnosis of Myelodysplastic Syndromes (MDS) following the 2008 WHO classification. Ann Hematol. 2013; 92: 19-24.
- Steensma DP. Dysplasis Has A Differential diagnosis: Distingushing Genuine Myelodysplastic Syndrome (MDS) From Mimics, Imitators, Copycats and Imposters.
- Blind men and an elephant-from Wikipedia, the free encyclopedia.
- Islam A, Catovsky D, Goldman JM, Galton DAG. Value of Long Core Biopsy in Detection of Discrete Bone Marrow Lesions. Lancet. 1979; 878.
- Islam A. A new bone marrow aspiration needle to overcome the sampling errors inherent in the technique of bone marrow aspiration. Journal of Clinical Pathology 1983; 36: 954-958.
- Tricot G, Boogaerts MA, De Wolf-Peeters C. The myelodysplastic syndromes: different evolution patterns based on sequential morphological and cytogenetic investigations. Br J Haematol. 1985; 59: 659-670.
- Vardiman JW, Brunning RD, Arber DA, Le Beau MM. Introduction and overview of the classification of the myeloid neoplasms. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC Press. WHO Classification of Tumours. 2008; 2: 18-30.
- Islam A. Bone marrow structure in human leukaemias: A histological study by plastic embedding techniques. 1982-Ph.D. thesis. Unlversity of London, UK.
- Islam A. Haemopoietic Stem Cell: A New Concept. Leukemia Research. 1985; 9: 1415-1432.
- Islam A, Catovsky D, Goldman JM, Galton DAG. Bone marrow biopsy changes in acute myeloid leukaemia I: observations before chemotherapy. Histopathology 1985; 9: 939-957.
- Islam A. Pattern of Bone Marrow Regeneration Following Chemotherapy for Acute Myeloid Leukemia. Journal of Medicine. 1987; 18: 108-122.
- Islam A, Henderson ES. Prediction of impending blast cell transformation in chronic granulocytic leukemia. Histopathology. 1988; 12: 633-639.
- Goldman JM, Johnson SA, Islam A, Catovsky D, Galton DAG. Haematological Reconstitution after Autografting for Chronic Granulocytic Leukaemia in Transformation: the Influence of Previous Splenectomy. British Journal of Haematology. 1980; 45: 223-231.
- Islam A, Catovsky D, Galton DAG. Histological Study of Bone Marrow Regeneration following Chemotherapy for Acute Myeloid Leukaemia and Chronic Granulocytic Leukaemia in Blast Transformation. British Journal of Haematology. 1980; 45: 535-540.
- Haase D, Germing U, Schanz J. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes of 2124 patients. Blood. 2007; 110: 4385-4395.
- Genovese G, Kahler AK, Handsaker RE. Clonal hematopoiesis and bloodcancer risk inferred from blood DNA sequence. N Eng J Med. 2014; 371: 2477-2487.
- Veryaskina YA, Titov SE, Kovynev IB. Prognostic Markers of Myelodysplastic Syndromes. Medicina (Kaunas). 2020; 56: 376-398.
- Olnes MJ, Sloand EM. Targeting Immune Dysregulation in Myelodysplastic Syndrome. JAMA. 2011; 305: 814-819.
- Valent P. Oligo-monocytic CMML and other pre-CMML states: Clinical impact, prognostication and management. Best Practice & Research Clinical Haematology. 2020; 33: 1-8.
- Valent P, Orazi A, Savona MR. Proposed diagnostic criteria for Classical Chronic Myelomonocytic Leukemia (CMML), CMML variants and pre-CMML conditions. Hematologica. 2019; 104: 1935-1949.
- Narang NC, Kotru M, Rao K. Megaloblastic Anemia with Ring Sideroblasts is not Always Myelodysplastic Syndrome. Turk J Haematol. 2016: 33: 358-359.
- Tenner S , Rollhauser C, Butt F. Sideroblastic anemia. A diagnosis to consider in alcoholic patients. Case Reports (Postgrad Med). 1992; 15: 147- 150.
- Nankivell BJ. Vitamin B6 Deficiency on Hemodialysis Causing Sideroblastic Anemia. Nephron. 1991; 59: 674-675.
- Knollmann-Ritschel BEC, Markowitz M. Lead Poisoning. Acad Pathol. 2017; 4: 2374289517700160.
- 10th Joyce Niblack Memorial Conference On Myeloproliferative Neoplasms, February 25 & 26, 2017. Presented by the MPN Education Foundation and the Mayo Clinic.
- Hong M, He G, The 2016 Revision to the World Health Organization Classification of Myelodysplastic Syndromes. J Transl Int Med. 2017; 5: 139–143.
- Jadersten M, Malcovati L, Dybedal I. Erythropoietin and granulocytecolony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol. 2008; 26: 3607-3613.
- Islam A. Hypoplastic acute myeloid leukemia in the elderly patient. A long term, continuous partial remission, and otherwise good health with low dose prednisone and G-CSF without chemotherapy. A case report. Clin Case Rep. 2019.
- Fenaux P, Platzbecker U, Mufti GJ. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N Engl J Med. 2020; 382: 140-151.
- Maximilian S, Zeidan AM. Lenalidomide use in myelodysplastic syndromes: Insights into the biologic mechanisms and clinical applications. 2017; 123: 1703-1713.