
Case Report
Ann Hematol Oncol. 2021; 8(1): 1323.
Severe Eosinophilia Associated with FIP1L1/PDGFRA Rearrangement Presenting with Yamaguchi Syndrome
Aamir Z¹*, Meraj F¹ and Hanif HM²
1Consultant Haematologist, The Indus Hopsital, Pakistan
2Department of Haematology, The Indus Hospital, Pakistan
*Corresponding author: Zunaira Aamir, Consultant Haematologist, The Indus Hospital, Plot C-76, Sector 31/5, Opposite Darussalam Society, Korangi Crossing, Karachi-75190, Pakistan
Received: December 20, 2020; Accepted: January 25, 2021; Published: February 01, 2021
Abstract
FIP1L1/PDGFRA is a rare genetic rearrangement, presenting most commonly as Chronic Eosinophilic Leukemia (CEL), but may also be associated with other myeloid and lymphoid neoplasms. The peripheral blood and bone marrow exhibit a striking eosinophilia, often associated with an increased number of mast cells on trephine biopsy. Tissue infiltration by eosinophils and release of cytokines from eosinophilic granules mediate multi-organ tissue damage. The tyrosine kinase inhibitor Imatinib has been shown to induce rapid and complete clinical and haematological responses in patients harboring the mutation. We present the case of a young patient with CEL associated with PDGFRA rearrangement, presenting with severe eosinophilia and evidence of multi-organ damage (cardiac, renal, endocrine and respiratory). The peripheral blood and bone marrow displayed a striking eosinophila, and FISH analysis for FIP1L1/PDGFRA revealed a positive fusion signal in 92% of the nuclei examined. Echocardiography showed left ventricular apical hypertrophy (Yamaguchi syndrome), which has previously not been reported in this subset of patients. He was managed with supportive care, along with low-dose imatinib (100mg/day initially), to which he achieved a rapid clinical and haematological response. Currently, five months from the initial diagnosis, he is doing well on low dose imatinib (100 mg) twice a week.
Keywords: Eosinophilia; Chronic eosinophilic leukemia; FIPL1/PDGFRA
Abbreviations
AEC: Absolute Eosinophil Count; CEL: Chronic Eosinophilic Leukemia
Background
Eosinophilia is a common clinical finding encountered in a wide range of diseases, including allergic disorders, infections, drugs and haematological malignancies. Haematological conditions associated with eosinophilia include lymphomas, acute myeloid leukemia, myelodysplastic syndromes, chronic eosinophilic leukemia NOS, myeloid/lymphoid neoplasms associated with rearrangement of PDGFRA, PDGFRB, FGFR1 and PCM1-JAK2 genes, and hypereosinophilic syndrome. In this report, we present the case of a young patient who presented with hyperleucocytosis (TLC 147.57x10^9/L) and severe eosinophilia (absolute eosinophil count 63.5x10^9/L), who was later found to have rearrangement in FIP1L1/ PDGFRA gene.
Myeloid/ Lymphoid neoplasms associated with PDGFRA rearrangement is a rare disease entity characterized by a cryptic deletion resulting in fusion of PDGFRA and FIP1L1 genes [1]. This rearrangement confers on haematopoietic precursor cells committment to eosinophilic differentiation [2]. The usual presentation is as Chronic Eosinophilic Leukemia (CEL), but patients may also present with acute myeloid leukemia and/or T lymphoblastic leukemia/lymphoma [3,4]. There is a marked predisposition to male sex (male: female ratio is 17:1) and median age of onset is in late 40s [3]. Tissue infiltration by eosinophils and release of cytokines from eosinophilic granules mediate multi-organ tissue damage. Hence, patients usually present with constitutional symptoms, accompanied by respiratory, cardiac, gastrointestinal and dermatological manifestations. The peripheral blood displays striking eosinophilia with mature morphology [3]. A range of morphological abnoramalities can be observed in eosinophils, including sparse granulation, cytoplasmic vacuolation and nuclear hyper- or hyposegmentation. The bone marrow is hypercellular with increased numbers of eosinophils and usually with a normal blast percentage. Bone marrow mast cells are often increased on trephine biopsy. The clonal eosinophils may show evidence of activation in terms of the immunophenotypic expression of CD23, CD25 and CD69.
The long-term prognosis of the disorder is still unclear, as its responsiveness to imatinib was not known until 2003 [5]. Nonetheless, if imatinib treatment is available and cardiac damage has not occurred, current evidence suggests the prognosis to be favorable3. Imatinib has been shown to induce rapid and complete clinical and haematological responses at doses ranging from 100-400 mg/day [6].
Case Presentation
A 30 year old male patient with no prior comorbids presented with complains of intermittent fever, progressive shortness of breath and pedal edema since 1 month. On general review, he had tachycardia (pulse rate 97/min), tachypnea (respiratory rate 20/min), postural hypotension and bilateral lower limb edema up to mid-thigh and hydrocele. Abdominal examination showed hepatosplemomegaly. Chest examination showed normal vesicular breathing in all areas with no cardiac murmur in areas of cardiac auscultation.
CBC showed bicytopenia and hyperleucocytosis (Hb 5.4 g/dl, TLC 147.57x10^9/L, Plt 44x10^9/L). Peripheral film examination revealed a striking eosinophilia (43% eosinophils) with mature morphology, leucoerythroblastic blood picture and no significant blast cells. Bone marrow examination showed a hypercellular marrow exhibiting 95- 100% cellularity and marked eosinophilic hyperplasia with all stages of differentiation. Scattered megakaryocytes were seen. There was no evidence of excess blasts, mast cells, significant dysplasia, granuloma, necrosis or metastatic disease in the available material. Reticulin stain showed Grade 1 myelofibrosis (loose network of reticulin with many intersections). On further workup, he was found to have prolonged INR (1.65), normal aPTT, deranged renal profile (urea 83mg/dl, creatinine 1.77 mg/dl, microalbuminuria 24.0mg/L) and liver function tests (total bilirubin 2.6mg/dl, direct bilirubin 1.64mg/dl, GGT 199U/L, ALT 634U/L, alkaline phosphatase 771U/L) and raised serum LDH (444 U/L). Serum TSH level was slightly low (0.347mIU/L; reference range 0.4-4.2 mIU/L), but free T3 and T4 levels were within normal limits. Ultrasound Abdomen showed significant hepatosplenomegaly (liver 19.5cm, spleen 18.3cm) and fatty infiltration of the liver. Chest X-ray showed two well defined densities in the right upper zone, resembling granulomas. Echocardiography showed left ventricular apical hypertrophy (Yamaguchi syndrome), and moderate tricuspid and mitral regurgitation. High sensitivity troponin I (103ng/L) and NT-proBNP (6403pg/ml) were raised. Thus, the patient had severe eosinophilia associated with evidence of multiorgan damage (cardiac, renal, endocrine and respiratory). He was initially managed with supportive care (hematinics, packed red cell transfusions, DVT prophylaxis, high protein diet, albumin replacement) and was started on Prednisolone 1mg/kg/day and Hydroxyurea 1g/day.
The following differential diagnosis were considered: chronic eosinophilic leukemia NOS, myeloid neoplasms with gene rearrangements (PDGFRA, PDGFRB, FGFR1 and PCM1-JAK2 genes), idiopathic hypereosinophilic syndrome and secondary causes of eosinophilia (infections, connective tissue disorders, etc.).
Bone marrow kayotyping revealed 46 chromosomes in all 20 cells counted and no significant abnormality. FISH analysis for FIP1L1/ PDGFRA revealed a positive fusion signal in 92% of the nuclei examined. The patient was thus diagnosed a case of Myeloid neoplasm with PDGFRA rearrangement presenting as chronic eosinophilic leukemia (WHO Classification 2017)³. He was later started on low dose imatinib 100mg/day. Two weeks after commencement of imatinib, the patient reported improvement in constitutional symptoms, but serial CBC monitoring showed pancytopenia (Hb 6.8g/dl, TLC 2.0x109/L with 2% Eosinophils, Platelets 54x109/L). His dose was reduced, and currently five months from the initial diagnosis, he is doing well on Imatinib 100 mg/day twice a week.
Discussion and Conclusion
Our patient is a case of chronic eosinophilic leukemia associated with PDGFRA rearrangement, presenting with severe eosinophilia and evidence of multi-organ damage. Several case reports and case series of patients with eosinophilia associated with FIP1L1/PDGFRA rearrangement, have been published from various countries [7-18]. In addition to the more common presentations, they have described the clinical features and outcome of this rare disease entity in paediatric patients, the occurrence of rare FIP1L1 breakpoint (located outside the usual breakpoint region) and atypical presentation of the disease with thrombotic thrombocytopenia purpura. In a case series comprising of 22 patients with mutated FIP1L1/PDGFRA, all patients were male with absolute eosinophil counts ranging from 1-110x109/L [17]. Most patients presented with constitutional symptoms, with evidence of organ involvement (skin, cardiac, pulmonary, gastrointestinal, neurologic) in a variable proportion of patients. Caridac involvement, characterized by congestive heart failure, left ventricular thrombus and characteristic echocardiographic findings, was noted in 7 (32%) of patients. Three patients were retrospectively identified, and they had not received imatinib treatment; in the remaining 19 patients, imatinib was started in 18 patients. Of the patients who received imatinib, complete haematological response was obtained in 17 patients; the remaining patient discontinued imatinib on account of intolerance, and was asymptomatic off-treatment, with an AEC of 1-1.5x109/L.

Figure 1: Bone marrow aspirate showing prominent eosinophilia and nuclear
hypersegmentation.

Figure 2: Low power (A) high power (B) views of bone marrow trephine
showing hypercelluar marrow with prominent eosinophilia.

Figure 3: Reticulin stain showing Grade 1 myelofibrosis (loose network of
reticulin with many intersections) (WHO classification, 2017³).
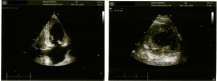
Figure 4: Echocardiogram showing apical myocardial hypertrophy.
In another case series comprising of 33 patients with CEL associated with PDGFRA rearrangement, cardiac involvement was present in 22% of patients, and manifested as endoomyocardial fibrosis, valvular abnormalities and mural thrombosis [18]. To our knowledge, this is the first case of apical hypertrophic cardiomyopathy (Yamaguchi syndrome) presenting in association with a clonal eosinophilia. Our patient displayed a rapid haematological response to imatinib in terms of reduction in his white cell count. It has been shown that a single weekly dose of imatinib, ranging from 100-200 mg/week is sufficient to maintain remission in patients with CEL harboring the FIP1L1/PDGFRA mutation [19].
Clinical Practice Points
• FIP1L1/PDGFRA is a rare genetic rearrangement, presenting most commonly as Chronic Eosinophilic Leukemia (CEL), but may also be associated with other myeloid and lymphoid neoplasms.
• Tissue infiltration by eosinophils and release of cytokines from eosinophilic granules mediate multi-organ tissue damage.
• The tyrosine kinase inhibitor Imatinib has been shown to induce rapid and complete clinical and haematological responses in patients harboring the mutation. A dose of 100-200 mg/week has been shown to be sufficient in maintaining remission.
Authors’ Contributions
ZA analyzed and interpreted the patient’s data and managed the patient. FM analyzed and interpreted the peripheral blood and bone marrow aspirate and trephine biopsy findings of the patient. HMH performed the bone marrow biopsy procedure, and was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
References
- Lancaster-Shorts KL, Chaffin J, Savage NM. Myeloid/Lymphoid Neoplams with abnormalities of PDGFRA. Atlas of Genetics and Cytogenetics in Oncology and Haematology. 2017.
- Fukushima K, Matsumura I, Ezoe S, Tokunaga M, Yasumi M, Satoh Y, et al. FIP1L1-PDGFRalpha imposes eosinophil lineage commitment on hematopoietic stem/progenitor cells. J Biol Chem. 2009; 284: 7719-7732.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. 2017.
- Metzgeroth G, Walz C, Score J, Siebert R, Schnittger S, Haferlach C, et al. Recurrent finding of the FIP1L1-PDGFRA fusion gene in eosinophiliaassociated acute myeloid leukemia and lymphoblastic T-cell lymphoma. Leukemia. 2007; 21: 1183-1188.
- Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003; 348: 1201-1214.
- Jovanovic JV, Score J, Waghorn K, Cilloni D, Gottardi E, Metzgeroth G, et al. Low-dose Imatinib Mesylate Leads to Rapid Induction of Major Molecular Responses and Achievement of Complete Molecular Remission in FIP1L1- PDGFRA-positive Chronic Eosinophilic Leukemia. 2007; 109: 4635-4640.
- Alshehri H, Alnomani M, Alghamdi M, Motabi I, Tailor I, Alshehry N, et al. An Intriguing Case of Eosinophilia with FIP1L1/PDGFRA Rearrangement Who Presented as Thrombotic Thrombocytopenic Purpura. Case Rep Hematol. 2019; 2019: 2820954.
- Lambert F, Heimann P, Herens C, Chariot A, Bours V. A case of FIP1L1- PDGFRA-Positive Chronic Eosinophilic Leukemia with a Rare FIP1L1 Breakpoint. J Mol Diagn. 2007; 9: 414-419.
- Rives S, Alcorta I, Toll T, Tuset E, Estella J, Cross NC. Idiopathic hypereosinophilic syndrome in children: report of a 7-year-old boy with FIP1L1-PDGFRA rearrangement. Journal of Pediatric Hematology/oncology. 2005; 27: 663-665.
- Chung KF, Hew M, Score J, Jones AV, Reiter A, Cross NC, et al. Cough and Hypereosinophilia Due to FIP1L1-PDGFRA Fusion Gene with Tyrosine Kinase Activity. Eur Respir J. 2006; 27: 230-232.
- Verdu J, Paz F. Chronic eosinophilic leukemia with FIP1L1-PDGFRA. Blood. 2013; 121: 1254.
- Srinivasan A, Scordino T, Baker A. Myeloid Neoplasm with Eosinophilia and FIP1L1-PDGFRA Rearrangement Treated With Imatinib Mesylate: A Pediatric Case With Long-term Follow-up. J Pediatr Hematol Oncol. 2019; 41: 334-335.
- Kim DS, Lee S, Choi CW. Loeffler endocarditis in chronic eosinophilic leukemia with FIP1L1/PDGFRA rearrangement: full recovery with low dose imatinib. Korean J Intern Med. 2018; 33: 642-644.
- Shin SY, Jung CW, Choi DC, Lee BJ, Kim HJ, Kim SH. Chronic eosinophilic leukemia with a FIP1L1-PDGFRA rearrangement: Two case reports and a review of Korean cases. Blood Res. 2015; 50: 58-61.
- Menif S, Omri H, Hafsia R, Rhomdane NB, Turki S, Meddeb B, et al. FIP1L1- PDGFRA positive chronic eosinophilic leukemia in Tunisian patients. Pathol Biol (Paris). 2007; 55: 242-245.
- Kumar AN, Sathyanarayanan V, Devi VL, Rajkumar NN, Das U, Dutt S, et al. FIP1L1-PDGFRA-positive chronic eosinophilic leukemia: a low-burden disease with dramatic response to Imatinib-a report of 5 cases from South India. Turk J Haematol. 2014; 31: 56-60.
- Pardanani A, Souza AD, Knudson RA, Hanson CA, Ketterling RP, Tefferi A. Long-term follow-up of FIP1L1-PDGFRA-mutated patients with eosinophilia: survival and clinical outcome. Leukemia. 2012; 26: 2439-2441.
- Qu SQ, Qin TJ, Xu ZF, Zhang Y, Ai XF, Li B, et al. Long-term outcomes of imatinib in patients with FIP1L1/PDGFRA associated chronic eosinophilic leukemia: experience of a single center in China. Oncotarget. 2016; 7: 33229- 33236.
- Helbig G, Stella-Holowiecka B, Majewski M, Calbecka M, Gajkowska J, Klimkiewicz R, et al. A Single Weekly Dose of Imatinib Is Sufficient to Induce and Maintain Remission of Chronic Eosinophilic Leukaemia in FIP1L1- PDGFRA-expressing Patients. Br J Haematol. 2008; 141: 200-204.