
Case Report
Ann Hematol Oncol. 2021; 8(1): 1325.
Additional Cytogenetic Aberrations Indicative of Poor Prognosis in AML with RUNX1/RUNX1T1 Rearrangement: Karyotype a Chromosomal Blueprint
Shetty D1*, Talker E1, Jain H1, Patil S1, Tembhare P2,3, Patkar N2,3, Nayak L3,4, Jain H3,4 and Sengar M3,4
1Department of Cancer Cytogenetics, Advanced Centre for treatment, Research and Education in Cancer (ACTREC), Tata Memorial Centre, India
2Department of Hematopathology, Advanced Centre for treatment, Research and Education in Cancer (ACTREC), Tata Memorial Centre, India
3Homi Bhabha National Institute (HBNI), Anushakti Nagar, India
4Department of Medical Oncology, Tata Memorial Hospital, India
*Corresponding author: Shetty D, Cancer Cytogenetic Department, Advanced Centre for Treatment Research and Education in Cancer, Tata Memorial Centre, India
Received: December 30, 2020; Accepted: February 02, 2021; Published: February 09, 2021
Abstract
AML is a highly heterogeneous disease with t(8;21)(q22;q22) as a frequently occurring aberration. While most studies show these patients to demonstrate a good response to standard chemotherapy, a lot of Indian and Western studies suggest otherwise. Trisomy 4 is a rare but a specific chromosomal abnormality in certain subtypes of AML. Although the significance of t(8;21) with trisomy 4 in AML remains uncertain, patients with this abnormality are typically associated with poor prognosis. Similarly, KIT mutations are also common with t(8;21) AML suggesting poorer outcomes. We report a case of AML with duplicated derivative 21 due to t(8;21), trisomy 4 and KIT mutation at baseline and an additional t(2;12) and ASXL2 mutation at relapse.
Keywords: AML; ASXL2; KIT; Trisomy 4
Introduction
The diagnosis of AML is based on the presence of ≥20% blasts in Bone Marrow (BM)/ Peripheral Blood (PB). Laboratory investigations including complete blood count, metabolic profile, bone marrow aspirate and biopsy (immunophenotype, cytochemistry), cytogenetics and mutation profiling further help risk-stratify these patients. Clinically, t(8;21)(q22;q22) is associated with good prognosis and a complete remission rate of 60%-70% (≤50 years) with infusional cytarabine and anthracycline therapy according to most trials [1]. Conversely, an increased relapse risk is seen with additional chromosomal abnormalities (ACA), thus, making ACA’s the most common determinants of prognosis in AML [2].
Presence of isolated trisomy 4 is rare (<1%) but is concomitant with t(8;21) AML [2]. So far, its prognostic significance remains disputed, partly due to its paucity as an isolated abnormality. Apparently, trisomy 4 does not have a significant impact on prognosis unless associated with KIT mutations which correlate with rapid disease progression [2]. According to the NCCN guidelines, KIT mutations often shift t(8;21) AML patients from low to intermediate risk [1]. Similarly, ASXL2 mutations and +der(21)t(8;21)(q22;q22) also confer poor outcomes [3].
We describe the clinicopathological findings of a RUNX1/ RUNX1TI positive AML case with mutated KIT, +4, +der(21)t(8;21) (q22;q22) at baseline, a rare t(2;12)(p13;p13) with ASXL2 mutation at relapse and highlight the prominence of cytogenetics.
Case Report
A 17-year old boy presented with fever (100.4°C), melena and body ache in December 2019. Peripheral blood examination showed anaemia, thrombocytopenia and leucocytosis. He had no comorbidities or familial history of cancer. Clinically he was suspected to have Acute Promyelocytic Leukemia (APML) and was started on arsenic trioxide. BM biopsy showed a hypercellular marrow with near-total replacement by blasts. Morphologic examination of the BM revealed 54% Myeloperoxidase (MPO) positive blasts with clonal mast cell population suggesting Acute Myeloid Leukemia (AML). Likewise, immunophenotype was also consistent with AML (Table 1). Biochemical examination showed low serum sodium and elevated serum CRP, globulin, bilirubin and LDH (Table 1).
![]()
Sr. No.
Laboratory Investigations
Report
1.
Bone marrow biopsy
Hypercellular marrow with near-total replacement by blasts, suggestive of acute leukemia
2.
Biochemistry
Serum CRP- 16.12mg/dL (high)
Serum Urea- 30.6 mg/dL (normal)
Serum Uric Acid-4.75mg/dL (normal)
Serum Creatinine-0.61mg/dL (normal)
Serum Sodium-133mmol/L(low)
Serum Potassium-4.76mmol/L (normal)
Serum Chlorides- 99.3 mmol/L (normal)
SerumBicarbonates-23.3 mmol/L(normal)
Serum Protein-7.89g/dL (normal)
Serum Albumin- 3.66g/dL (normal)
Serum Globulin-4.23g/dL (high)
Serum Alk.Phosphatase- 155U/L (normal)
Total Bilirubin-1.21mg/dL (high)
Serum AST- 33U/L (normal)
Serum ALT- 44U/L (normal)
Serum LDH- 2012 U/L (high)
Serum Calcium-9.37mg/dL (normal)
Serum Phosphorus-4.24mg/dL(normal)3.
Haematology
Haemoglobin- 8.20g/dL(low)
RBC count- 2.41x 10e12/L (low)
Platelets- 24.00x 10e9/L (low)
WBC count- 16.05x 10e9/L (high)4.
Flow cytometry
CD 45, CD 38, CD25, CD 15, CD 123, CD 13, CD 19– dim negative
CD 20, CD 11b, CD 36,CD 1a, CD 5, CD 2, CD 10, CD 22, CD73, CD 16, CD 11c, sCD 3, CD 7, TCRGD, CytoCD3, CD86 , CD 304, CD 14, CD 163, CD 4, CD 8, CD42b– negative
HLA–DR, CD 117– variable
CD 56– bright
cCD 79a– small subset
CD 34– moderate
CD 33– dim moderate
AMPO– dim positive
Blasts– 54%
MPO– Positive5.
Cytogenetics
At baseline
Karyotype:48,XY,+4,+der(21)t(8;21)(q22;q22),t(8;21)(q22;q22)[10]/46,XY[10]
FISH: RUNX1/RUNX1TI fusion–97% cells (3F1R1G)
Follow–up at 2 months
Karyotype:46,XY[20]
FISH: Negative for RUNX1/RUNX1TIfusion
Follow–up at 10 months
Karyotype:48,XY,t(2;12)(p13;p13),+4,+der21,t(8;21)(q22;q22)[19]/46,XY[1]FISH: RUNX1/RUNX1TI fusion– 95% cells (3F1R1G)5.
Next–generation sequencing
At baseline
Positive for a missense (p.D816V) mutation in the exon 17 of the KIT gene. Variant Allele Frequency: 48.58%
Follow–up at 2 months
RQ–PCR: RUNX1–RUNX1T1: POSITIVE
Number of RUNX1–RUNX1T1 fusion copies: 9462
Number of ABL1 copies: 2448000
Normalised copy number: 0.38
Follow–up at 10 months
- Missense mutation in the exon 17 ofthe KIT gene
- Stopgain mutation in ASXL2 gene
6.
MRD
@ 2months: 0.012%
@10 months: RelapseAbbreviations: CRP:C–Reactive Protein; AST: Aspartate Aminotransferase; ALT: Alanine Aminotransferase; LDH: Lactate Dehydrogenase; RBC: Red Blood Cells; WBC: White Blood Cells; CD: Clusters of Differentiation; HLA–DR– Human Leukocyte Antigen– DR Isotype; MPO: Myeloperoxidase; F: Fusion; R: Red; G: Green
Table 1: Laboratory investigations of the patient at baseline, 1st follow-up and relapse.
Fluorescence In-Situ Hybridization (FISH) and GTG-banding was performed as part of routine cytogenetic work-up. Unstimulated BM cultures were set-up (direct and overnight) using RPMI 1640 media. Colcemid arrested metaphases were treated with hypotonic 0.075mol/L KCl and fixed with Carnoy’s fixative. Giemsa banded metaphases were analysed using the Carl Zeiss Axioimager.Z2 microscope with MetaSystems analysis software- IKAROS and karyotype- 48,XY,+4,+der(21)t(8;21)(q22;q22),t(8;21)(q22;q22) [10]/46,XY[10] (Figure 1) was established according to ISCN 2016 [4]. The FISH was performed for standard AML panel including LSI RUNX1/RUNX1T1 dual colour dual fusion probe, LSI MLL break apart probe, LSI CBFβ break apart probe, LSI BCR/ABL1 dual colour dual fusion probe, LSI TP53/CEP17 deletion probe, CEP 8, LSI 5p15/q31/q33 triple colour deletion probe and LSI 7q22/q36/CEP7 triple colour deletion probe (MetaSystems, Altlussheim, Germany). FISH analysis was done using the Carl Zeiss Axioimager.Z2 with MetaSystems analysis software- ISIS. Interphase FISH using LSI RUNX1/RUNX1T1 showed 97% cells with 3F1R1G pattern (Figure 1) while metaphases revealed RUNX1/RUNX1TI fusion on derivative 8, derivative 21 and duplicated der (21) along with 1 red and green signal on normal chromosomes 8 and 21 respectively (Figure 1). No other abnormalities were detected by FISH. Whole chromosome paints for chromosome 2 and 12 (MetaSystems, Altlussheim, Germany), CEP 4 and RUNX1/RUNX1T1 probe were used in combination to confirm t(2;12), t(8;21) and +4 (Figure 1). Next-Generation Sequencing (NGS) was positive for a missense [p.(Asp816Val)] mutation in exon 17 of the KIT gene (Variant Allele Frequency: 48.58%). The patient was started on 3+7 induction (Daunomycin-600mg/m2; Cytarabine- 100mg/m2/day). Clinical examination before initiation of induction showed no enlarged nodes, spleen or liver. The patient, however, experienced low-grade fever, nausea and neck pain through the course of therapy. Post induction blood for galactomannan test and microbial cultures was negative. NCCT thorax revealed no significant abnormality, the liver and spleen were non-palpable and the 2D ECHO was normal. Post induction Minimal Residual Disease (MRD) at 1st follow-up was 0.012% with 9% blasts and karyotype and FISH were normal. The patient was initiated on HiDAC (3 cycles).
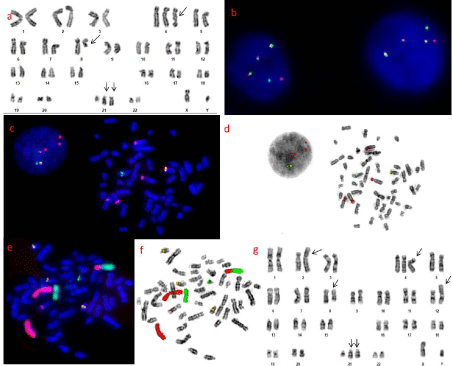
Figure 1: Chromosomal abnormalities at baseline and at relapse.
a. Karyotype at baseline: 48,XY,+4,+der(21)t(8;21)(q22;q22),t(8;21)(q22;q22)
[10]/46,XY[10]
b. Interphase FISH using RUNX1/RUNX1T1 dual fusion probe showing
3F1R1G signals
c. Metaphase FISH with ETV6 break apart probe and CEP 4 showing 1F4R1G
signals; 1F on normal chromosome 12, 3 red signals on the centromere of
chromosome 4 suggesting trisomy 4, 1 red signal on der(2) and 1G signal on
der(12) suggesting t(2;12)
d. Reverse DAPI image of Figure. 1c
e. Metaphase and interphase FISH with a cocktail of WCP 2 (red) and 12
(green) along with RUNX1/RUNX1T1 dual fusion probe confirming t(2;12)
and RUNX1/RUNX1T1 fusion with duplication of der(21)t(8;21)(q22;q22)
f. Reverse DAPI image of Figure. 1e
g. Karyotype at relapse: 48,XY,t(2;12)(p13;p13),+4,+der21,t(8;21)(q22;q22)
[19]/46,XY[1]
After ten months, he relapsed with 88% BM blasts. FISH showed RUNX1/RUNX1T1 fusion with +der(21) in 95% cells. Cytogenetics showed karyotype- 48,XY,t(2;12)(p13;p13),+4,+der(21)t(8;21) (q22;q22)[19]/46,XY[1] (Figure 1). NGS revealed the presence of a missense mutation in exon 17 of the KIT gene and a stopgain mutation in ASXL2 gene [p.(Glu1172Ter); Variant allele frequency: 63.20%]. He was initiated on the CLAG regimen in view of relapse, however, the patient died in October 2020.
Discussion
The t(8;21)(q22;q22) a frequent chromosomal abnormality (~5% AML and 10% AML-M2) among younger AML patients is concomitant with loss/inactivation of sex chromosomes, deletion of 9q12~q23 (20%) and trisomies 4 and 8 (6-10%) [3]. Although RUNX1/RUNX1T1 is favourable-risk cytogenetics, associated additional abnormalities shift the prognosis towards high-risk. Trisomy 4 with t(8;21) shows unfavourable clinical outcome mostly due to concomitant KIT mutation which roots the adverse effects of the disease [2]. According to literature, KIT mutations occurs solely at a frequency of 32% while 10% show t(8;21) and/or +4 [5].
Our patient had clinical features suggestive of APML however, cytogenetic analysis showed t(8;21) and immunophenotype suggested AML. Cytogenetically, +der(21) was observed. According to literature, duplication of ETO-AML1 corresponds to poor outcomes, regardless of its presence at baseline or relapse [3]. Our patient also had a KIT mutation at baseline. He initially showed remission, but the disease relapsed after 10 months. A similar case, suggesting the poor outcome of +4 with t(8;21) was reported in a Malaysian woman with AML who died soon after diagnosis [6].
At relapse, a rare translocation- t(2;12)(p13;p13) involving ETV6 was seen along with ASXL2 mutation and baseline abnormalities. Although the ETV6 gene has ~40 known partners, its occurrence in myeloid neoplasms is rare (0.5%) and reports suggest that ETV6 does not play a major role in AML pathogenesis [7]. In literature, there are very few reports of AML with t(2;12)(p13;p13); Walker et al. (2013) have reported two cases, both of which died within six months of diagnosis [8]. Likewise, Hagiwara et al. (1998) have also reported t(2;12)(p13;p13) at relapse [9]. ASXL2 mutations have been identified in ~16%-22% RUNX1/RUNX1T1 AML and their synergistic presence is associated with a higher 3-year cumulative incidence of relapse (36%) as against ASXL1/2 (25%) wild-type counterparts [10].
Although the exact mechanism of cytogenetic evolution is not completely understood, it must be disease-specific. It is believed that the t(8;21) initiates the disease, followed by activation of the KIT pathway and increased gene-dosage of aberrant KIT due to trisomy 4 which promotes disease progression [5].
Conclusion
AML with +der (21)t(8;21), +4 and/or KIT mutations should be more precisely stratified and closely monitored for appropriate risk-adapted therapy and predicting relapse risk. Besides, this case exemplifies the prominence of conventional cytogenetics and FISH in confirming the diagnosis and in prognostication.
References
- Haferlach C, Bacher U, Schnittger S, Alpermann T, Zenger M, Kern W, et al. ETV6 rearrangements are recurrent in myeloid malignancies and are frequently associated with other genetic events. Genes chromosome cancer. 2011; 51: 328-337.
- Hagiwara S, Yuo A, Miwa A, Takezako N, Hirano N, Togawa A. A rare atypical myeloproliferative disorder-like hemopathy with marked dysplasia, peripheral dominant myeloblast proliferation and extramedullary hematopoiesis was converted into typical acute myeloid leukemia with an interval of complete hematological remission. Int J Hematol. 1998; 67: 411-416.
- Jahn N, Agrawal M, Bullinger L, Weber D, Corbacioglu A, Gaidzik VI, et al. Incidence and prognostic impact of ASXL2 mutations in adult acute myeloid leukemia patients with t(8;21)(q22;q22): a study of the German-Austrian AML Study Group. Leukemia. 2017; 31: 1012-1015.
- McGowan-Jordan J, Simons A, Schmid M. An International system for human cytogenomic nomenclature (ISCN) 2016. Basel: Karger. 2016.
- Mikulasovicha M, LeBlanca A, Scalisea A, Manwanic D, Keyznerb A, Najfelda V. Duplication and triplication of der(21)t(8;21)(q22;q22) in acute myeloid leukemia. Can Genet Cytogenet. 2009; 188: 83-87.
- National Comprehensive Cancer Network clinical practice guidelines in oncology (NCCN Guidelines): acute myeloid leukemia. Version 1. 2018.
- Phan CL, Ong TC, Chang KM, Zubaidah Z, Puteri Jamilatul NMB. Concomitant t(8;21) and Trisomy 4 in a Patient with Acute Myeloid Leukemia (AML). Med Health. 2010; 5: 45-48.
- Shimada A, Taki T, Tabuchi K, Tawa A, Horibe K, Tsuchida M, et al. KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood. 2006; 107: 1806-1809.
- Trivedi PJ, Patel PS, Brahmbhatt MM, Patel BP, Gajjar SB, Iyer RR, et al. A case of acute myeloid leukemia-M2 with trisomy 4 in addition to t(8;21). Indian J Hum Genet. 2008; 14: 20-22.
- Walker A, Mrózek K, Kohlschmidt J, Rao K, Pettenati M, Sterling L, et al. New Recurrent Balanced Translocations in Acute Myeloid Leukemia and Myelodysplastic Syndromes: Cancer and Leukemia Group B (CALGB) 8461. Genes Chromosomes Cancer. 2013; 52.