
Case Report
Ann Hematol Oncol. 2021; 8(3): 1333.
Unusual Manifestations of Paraprotein-Associated Hematological Diseases: Report of 2 Cases and Literature Review
Karakatsanis S¹*, Papadatos SS¹, Polyzos D¹, Gravani A¹, Syrigos N¹, Tsakania A², Kanellis G³, Marinos L³ and Papanikolaou A³
¹Department of Internal Medicine, National and Kapodistrian University of Athens, Greece
²Department of Blood Transfusion, “Sismanogleio” General Hospital, Greece
³Department of Hematopathology, Evangelismos General Hospital, Greece
*Corresponding author: Karakatsanis S, Department of Internal Medicine, National and Kapodistrian University of Athens,152 Mesogeion Avenue, 115 27, Athens, Greece
Received: February 04, 2021; Accepted: March 04, 2021; Published: March 11, 2021
Abstract
With this review we firstly describe uncommon manifestations of paraprotein-associated hematological diseases in 2 patients presenting with hemoptysis. The differential diagnosis in both cases included qualitative and quantitative platelet abnormalities, infectious diseases, malignancies, vasculitis, collagen diseases and blood disorders. Among other interventions undertaken, bone marrow biopsy provided evidence of a primary hematological disease while serum protein electrophoresis and immunofixation indicated the presence of a paraprotein. As a “take-away” lesson from the literature review performed on the basis of these cases the bleeding diathesis that is occasionally associated with paraproteinemia should be pointed out, irrespectively of the primary hematological condition. Thus, while investigating a patient with bleeding diathesis, at least a serum protein electrophoresis should be among the requested laboratory tests. Moreover, clinical vigilance is required in order to recognize other unusual manifestations of not so unusual hematological abnormalities.
Keywords: Bleeding; Hemoptysis; Leukoerythroblastic reaction; Lymphadenopathy; Organomegaly; Paraprotein
Abbreviations
A/G Ratio: Albumin to Globulin Ratio; AL: Systematic Immunoglobulin Light Chain Amyloidosis; AVWS: Acquired von Willebrand syndrome; BAL: Bronchoalveolar Lavage; BMB: Bone Marrow Biopsy; CLL: Chronic Lymphocytic Leukemia; CrCl: Creatinince Clearance; DAH: Diffuse Alveolar Hemorrhage; DAT: Direct Antiglobulin Test; ESR: Erythrocyte Sedimentation Rate; F: Factor; GGO: Ground-Glass Opacity; HBsAg: Surface Antigen of the Hepatitis B Virus; HBV: Hepatitis B Virus; Ig: Immunoglobulin; IGRA: Interferon-γ Release Assay; IMWG: International Myeloma Working Group; ITP: Immune Thrombocytopenia; LDH: Lactate Dehydrogenase; LPD: Lymphoproliferative Disease; MBL: Monoclonal B Lymphocytosis; MDRD: Modification of Diet in Renal Disease; MGUS: Monoclonal Gammopathy of Undetermined Significance; MM: Multiple Myeloma; MZL: Marginal Zone Lymphoma; PB: Peripheral Blood; PCD: Plasma Cell Disorder; PET/ CT: Positron Emitting Tomography Combined with Low-Dose Computed Tomography; POEMS: Polyneuropathy, Organomegaly, Endocrinopathy, Multiple Myeloma And Skin Changes; RBC: Red Blood Cell; R-ISS: Revised International Staging System; RLL: Right Lower Pulmonary Lobe; RUL: Right Upper Pulmonary Lobe; SMZL: Splenic Marginal Zone Lymphoma; TST: Tuberculin Skin Test; TTP: Thrombotic Thrombocytopenic Purpura; vWF: von Willebrand Factor
Introduction
Multiple Myeloma (MM) is the second commonest hematological malignancy in the Western world since it accounts for 1% of all cancers and 10% of all hematological malignancies [1]. Most patients have characteristic clinical and laboratory findings, including bone pain with or without pathologic fractures, anemia and/or weakness, infections, hypercalcemia and renal failure [2]. On the other hand, Monoclonal Gammopathy Of Undetermined Significance (MGUS) is a much commoner premalignant, clonal Plasma Cell Disorder (PCD), present in 3% to 4% of the general population older than 50 years and characterized as well by the presence of a Monoclonal (M)- protein (or paraprotein), but lacks by definition MM clinical findings [3]. Paraproteins may be found among patients with rarer PCDs as well as lymphoplasmacytic malignancies and other low-grade B-cell non-Hodgkin lymphomas with plasmacytic differentiation [2,4,5]. The following 2 cases illustrate that paraproteins, irrespective of their association with benign or malignant blood disorders, can disrupt normal coagulation through various mechanisms, and therefore patients with bleeding diathesis should also be evaluated for the presence of a monoclonal protein. What is more, physicians should also be aware of other unusual manifestations of the aforementioned conditions.
Case Presentation
Case report 1
A 52-year-old female accountant, current smoker, presented at our Emergency Department because of 2 episodes of hemoptysis during the last 24 hours, yet her examination from the attending pulmonologist did not reveal a specific thoracic disease, and she was further referred to our Hematology Department because of the alleged hemoptysis in combination with thrombocytopenia that was found in her full blood count.
A comprehensive systems’ review came up with episodes of spontaneous bruising and epistaxis for the last 3 months, while the patient also mentioned increased blood loss during her last menstrual cycle. No other systemic or otherwise symptoms were reported. The patient’s personal history included only arterial hypertension under treatment and appendicectomy in young age, while her physical examination was remarkable for stage 1 obesity, bilateral tonsil hyperplasia, mild conjunctival pallor and a palpable spleen. Laboratory results included leukocytosis with absolute lymphocytosis, severe macrocytic anemia, mild thrombocytopenia (>50,000/μlt), high erythrocyte sedimentation rate (ESR), a low Albumin to Globulin (A/G) ratio, absent haptoglobins, and liver function tests abnormalities combined with positive surface antigen of the Hepatitis B Virus (HBsAg). The rest of her microbiology results were negative. HBV-DNA was subsequently sent and came up positive (2.12x107IU/ml or 1.03x108 copies/ml). Of note, routine coagulation tests (prothrombin time, activated thromboplastin time, fibrinogen) were all normal.
Peripheral Blood (PB) smear examination provided evidence of a leukoerythroblastic reaction, an absence of lymphocytosis surprisingly, macrocytosis, fine basophilic stippling, rouleaux formation and the presence of Red Blood Cell (RBC) agglutination, spherocytes, but no fragmented RBCs, and a limited number of plasma cells (Figure 1). Computed tomography scan revealed massive splenomegaly (measured dimensions were 13.7x14x10 cm³), hepatomegaly (cephalocaudal diameter measured at 18.7 cm), intraabdominal lymphadenopathy, multiple large uterine fibroids (with maximum diameter ~8cm) and a Ground-Glass Opacity (GGO) of the Right Upper Pulmonary Lobe (RUL).
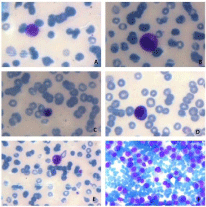
Figure 1: Peripheral blood smear (A-E) showing leukoerythroblastic reaction
and bone marrow aspirate smear (F) showing plasma cell infiltration. A: An
erythroblast; B: A plasma cell; C: A promyelocyte; D: A nucleated red blood
cell; E: A myelocyte; F: Bone marrow plasma cell infiltration.
At that time, based on the aforementioned clinical, laboratory and imaging findings, in our differential diagnosis we included qualitative and quantitative platelet abnormalities, other bleeding disorders and bone marrow suppression and/or infiltration due to infectious agents, malignant disorders, collagen and hematological diseases. Laboratory results were negative for the presence of antiphospholipid antibodies, her Tuberculin Skin Test (TST) and Interferon-γ Release Assay (IGRA) were also negative, while the rest of the autoimmune panel did not reveal a specific abnormality apart from low C3 and even lower C4 levels. On the other hand, serum ferritin levels were within normal limits. Bone Marrow Biopsy (BMB) was then undertaken while at the same time a bone marrow smear was examined, immunophenotyping of the bone marrow aspirate was requested and a bone marrow sample was sent in search for PML/RARa translocation due to the bleeding diathesis and the occasional presence of blasts in the PB smear. The results were negative for the presence of acute leukemia, however an increased Β2 microglobulin value at 11.4mg/lt was noted while a quantitative serum immunoglobulin test indicated an IgG of 9,000mg/dL with associated immunoparesis. Serum and urine immunofixation detected the presence of a IgGλ paraprotein, while her Positron Emitting Tomography combined with Low-Dose Computed Tomography (PET/CT) was unremarkable, just like her portal venous Doppler imaging. Finally, the patient’s result of BMB was only consistent with the presence of an IgGλ MM, since a bone marrow infiltration by monoclonal cIgGλ plasma cells by 80% was evident. The patient’s karyotype came back normal but her Lactate Dehydrogenase (LDH) value was elevated, so her Revised International Staging System (R-ISS) stage was calculated as III. The patient asked then to be referred to a tertiary Hematology Clinic with expertise in treating MM patients, in order to receive the appropriate treatment.
Case report 2
A 76-year-old male retired high-school teacher, former smoker (he had quit 30 years ago and had a smoking history of 10 pack-years), was referred to our Hematology Department because of a serum paraprotein found during evaluation of episodes of hemoptysis since 5 months ago and worsening the last 24 hours. A comprehensive systems’ review was otherwise unremarkable. The patient’s personal history included coronary artery disease with associated heart failure and his physical examination was remarkable for stage I obesity, mild hypoxia (oxygen saturation 92%) and bibasilar (but mostly on the right) coarse crackles. Our patient had been first admitted in a Pulmonary clinic due to his main symptom and abnormal findings on chest CT, i.e., crazy paving appearance and alveolar infiltrates in his Right Lower Pulmonary Lobe (RLL) with concomitant GGOs (commented on CT report as probable alveolar hemorrhage), an adjacent nodular lesion ~1cm in contact with the oblique fissure, intrathoracic lymphadenopathy (with a reported maximum diameter of 12mm), and 2 subpleural nodules with a diameter of ~2-3 mm, possibly representing granulomas. Abnormal laboratory results included absolute lymphocytosis, mild normocytic anemia, high ESR, moderate chronic kidney disease [Creatinince Clearance (CrCl) ~65ml/min and estimated glomerular filtration rate using the MDRD formula ~52ml/min/1.73 m2] with a normal urine sediment examination, a positive d-dimer test (but without any evidence of pulmonary embolism on CT pulmonary angiogram), a positive Direct Antiglobulin Test (DAT), raised levels of rheumatoid factor and low levels of complement, and a low A/G ratio. Therefore, in our differential diagnosis we included vasculitis, connective tissue disease, pulmonary alveolar proteinosis, lower respiratory tract infection, malignancy and PCD.
Bronchoscopy and Bronchoalveolar Lavage (BAL) were undertaken and revealed Diffuse Alveolar Hemorrhage (DAH). PB smear examination provided evidence of absolute lymphocytosis with small, mature lymphocytes and rouleaux formation. His PB immunophenotyping was not compatible with Chronic Lymphocytic Leukemia (CLL) diagnosis and a quantitative serum immunoglobulin test indicated raised IgG and IgM, hence at that time our differential also included a Lymphoproliferative Disease (LPD) with plasmacytic differentiation. The Bence-Jones protein urine test was negative but serum immunofixation revealed the presence of a IgGκ paraprotein. Whole-body low-dose CT was without findings and a BMB was then undertaken, which was consistent either with the presence of Marginal Zone Lymphoma (MZL) plus a biclonal plasmacytic differentiation (monoclonal cIgGκ as well as a small percentage of monoclonal cIgMκ plasma cells were observed), or MZL plus its plasmacytic component plus MGUS. PET/CT showed unhomogeneous hypermetabolism implying a bibasilar, reticular interstitial pattern which was attributed to the underlying LPD. A new CT scan showed a marginal improvement regarding the lung parenchyma but some lymph nodes had increased in size. Pending further therapeutic decisions, the patient was referred to a specialized Hematology laboratory so that his bleeding diathesis would be further investigated, since his platelet count and routine coagulation tests were all normal.
Discussion
Initially, we report herein a patient with IgGλ MM and atypical clinical presentation as well as laboratory results, including massive splenomegaly, hepatomegaly, intraabdominal lymphadenopathy, leukoerythroblastic reaction, spherocytes and absent haptoglobins. Nevertheless, her most unsettling symptom was hemoptysis, a manifestation of her bleeding diathesis (in spite of her mild thrombocytopenia and normal coagulation tests) that had also expressed itself with spontaneous bruising, epistaxis, and increased blood loss during menstruation. The aforementioned RUL GGO might have been another evidence of the patient’s bleeding diathesis, representing a possible, isolated site of alveolar hemorrhage. Unfortunately, we did not have a chance to further evaluate this radiological finding with bronchoscopy and BAL.
Our second patient was diagnosed with a low-grade LPD ± MGUS, complicated by hemoptysis due to DAH as it was proven by BAL. His bleeding diathesis did not have as well a straightforward explanation, since his platelet count and coagulation tests were normal. Interestingly, in both patients an IgG paraprotein was evident, first hinted by a high ESR and low A/G ratio and then confirmed with serum electrophoresis and immunofixation.
Certain MM patients or individuals with other PCDs have an increased risk of developing a coagulation disorder and subsequent bleeding, in addition of course to those patients that develop severe thrombocytopenia. In general, MM patients may be exposed to a high risk of both spontaneous and peri-interventional bleeding, since about 15% of MM patients present with a bleeding diathesis [6,7]. The same holds true for MGUS, since monoclonal gammopathy may have various manifestations, including bleeding diathesis, related to adsorption of biologically active molecules on clonal cells or aggregated paraprotein [8]. However, clinically important bleeding complications such as hemoptysis among PCD patients are not usually seen [7].
The relevant mechanisms of coagulation abnormalities in MM are complex and reflect an abnormal interaction between clonal plasma cells, inflammatory pathways and M proteins on one side and platelets, von Willebrand Factor (vWF), coagulation factors and the vasculature on the other [7,9,10]. The literature is rich mostly with case reports commenting on MM patients with Acquired von Willebrand syndrome (AVWS) [11-20], acquired haemophilia A [19,21-25], dysfibrinogenemia due to antibodies directed against fibrinogen [26-29] or due to defects in fibrin polymerization [30- 33], excessive fibrinolysis [34,35], increased plasma viscosity caused by high protein concentration in the blood [36-38], the presence of cryoglobulins [39], as well as specific coagulation factors’ deficiencies, such as Factor V (FV) and FX deficiency [40-42]. Moreover, there are reports of an acquired heparin-like coagulation inhibitor [10,43,44] and Thrombotic Thrombocytopenic Purpura (TTP) [45,46], while impaired platelet function has also been under the spotlight [9,20,47]. Similar mechanisms have been described in individuals with MGUS, i.e., AVWS [48-63], presence of cryoglobulins [64,65] or acquired coagulation inhibitor [66], and Immune Thrombocytopenia (ITP) [67], while only one case series has been published up until now (in French) regarding patients with MZL, paraprotein and bleeding diathesis due to AVWS [68], as well as 2 case reports [69,70] regarding patients with splenic MZL (SMZL), with one of the patients also having a biclonal gammopathy [69]. There is also a case report of a MZL patient with spontaneous hematomas due to acquired FX deficiency [71] and a case report of a patient with acquired hemophilia A and a circulating Monoclonal B Lymphocytic population (MBL) with a marginal zone phenotype [72]. Lastly, bleeding diathesis among systematic immunoglobulin light chain (AL) amyloidosis patients is also attributed to AVWS, activation of the fibrinolytic system, decreased synthesis of coagulation factors in advanced liver disease, as well as amyloid infiltration of blood vessels [10,16,41,73,74]. The International Myeloma Working Group (IMWG) has issued recommendations for prophylactic and therapeutic anticoagulation in MM patients [75] but interestingly enough, not for a possible bleeding diathesis among them or other patients with PCD. Moreover, it is currently unknown if only laboratory-evident coagulopathy has any prognostic role [41].
Mass disease or organomegaly in MM patients is usually due to extramedullary plasmacytomas or amyloidosis and found in approximately 10% of patients [1]. Splenomegaly and hepatomegaly are thought to be rare clinical findings in patients with MM but without amyloidosis, with respective incidence of <1% and 4-5 % in newly-diagnosed patients [76-80]. Patients that develop MM after having been diagnosed with Gaucher disease could also have organomegaly (as well as lymphadenopathy) since before their MM diagnosis [81,82] while there is also a recent case report describing a MM patient with acquired hemophagocytic lymphohistiocytosis as initial manifestation of MM, and thus splenomegaly [83]. It should be kept in mind that organomegaly is much commoner among patients with AL and rare plasma cell dyscrasias such as POEMS (Polyneuropathy, Organomegaly, Endocrinopathy, Multiple myeloma and Skin changes) syndrome [84,85], while patients with primary plasma cell leukaemia also have a higher incidence of hepatosplenomegaly and lymphadenopathy compared to MM patients [86]. Regarding lymphadenopathy among MM patients, case reports have been infrequently published in the literature [87-91], while not so current patient series have reported an incidence of ≤1% [76,78,80]. Finally, to the best of our knowledge, leukoerythroblastic reaction in a MM patient has been reported only once before [79], while there is no report regarding the presence of spherocytes in MM. Finally, with regard to MM and low or absent haptoglobins, haptoglobin levels have been found actually increased among MM patients [92,93].
The common symptom of our 2 patients was their bleeding diathesis (manifested mainly as hemoptysis), while they also shared low complement levels, the presence of an IgG paraprotein and a seemingly intact coagulation system (since their standard coagulation assays were normal). So, it would not be inappropriate to attribute their common clinical picture to their common and bibliographically well-established laboratory finding, e.g., their IgG paraprotein, despite the fact that the last one has been reported to have the lowest bleeding rate [94-97].
Additional considerations regarding our first patient were, to start with, that her bleeding diathesis could be ascribed in part also to her mild thrombocytopenia, which in turn could be also attributed to a chronic, immune-active Hepatitis B Virus (HBV) infection (positive HBsAg and high viral load) and hypersplenism (either due to splenomegaly or not), yet our patient had no evidence of impaired hepatic synthetic function and decompensated cirrhosis, second, that at the time of her diagnosis she might also have an overt hemolysis due to the observed massive splenomegaly, hence justifying the absent haptoglobins and the presence of leukoerythroblastic reaction, basophilic stippling and spherocytes, third, that the constellation of organomegaly, lymphadenopathy and thrombocytopenia might have been suggestive of an undiagnosed Gaucher disease, yet the lack of bone marrow infiltration by Gaucher cells is against such possibility, and four, that the patient might have congenital ahaptoglobinemia (or anhaptoblobinemia) [98], thus the undetectable haptoglobin level. Finally, her low complement levels could be indicative of vasculitis, or the patient’s hemoptysis could be independent of her paraproteinemia and only due to a local cause that we did not have the chance to investigate further. Yet, her generalized bleeding diathesis and bleeding pattern (mostly her late onset bruising, epistaxis and heavy menstrual bleeding) combined with her normal coagulation studies point to an AVWS [99].
Regarding our second patient, an apparent consideration due to his raised levels of rheumatoid factor and low levels of complement was the possibility of vasculitis-driven hemoptysis, however his urine increment came back normal. Another concern we have is that his bleeding symptoms were limited only in his respiratory tract. His specialized coagulation investigation results are still pending but again, based on his normal coagulation studies, we believe that his diagnosis possibly is AVWS due to IgG paraprotein. We acknowledge however that hemoptysis is rare among patients with AVWS or paraproteinemia (excluding cases of pulmonary plasmacytomas or other PCD localized involvement), at least according to current literature [100-105] (Table 1).
![]()
Author (year of publication)
Article category
No of patients
Sex
Age (years)
Other manifestations
Primary disease diagnosis
Diagnosed hemostatic abnormality
Reference
Bolaman (2008)
Letter to the editor
1
Female
49
Anemia
MM
NR
[100]
Morita (2010)
Case report
1
Female
84
Dyspnea, hematuria, progressive anemia, malaise, cough
MM
NR
[101]
Szeto (2013)
Case report
1
Male
67
Cough, progressive dyspnea, malaise
MM
NR
[102]
Coucke (2014)
Case report
1
Male
36
Anemia
WM
AVWS
[103]
Clay (2018)
Case report
1
Female
48
No
Systemic AL amyloidosis with cardiac involvement
NR
[104]
Uwingabiye (2018)
Case report
1
Male
59
Renal failure, anemia, thrombocytopenia, hemothorax
PCL
anti-factor XI antibodies
[105]
Present study
Case series
2
1 female
52
Epistaxis, spontaneous bruising
MM
NR
1 male
76
Mild dyspnea
MZL plus a biclonal plasmacytic differentiation or MZL plus its plasmacytic component plus MGUS
NR
AL: Immunoglobulin Light-Chain Amyloidosis; AVWS: Acquired Von Willebrand Syndrome; BMA: Bone Marrow Aspiration; BMB: Bone Marrow Biopsy; LPL: Lymphoplasmacytic Lymphoma; MGUS: Monoclonal Gammopathy Of Undetermined Significance; MM: Multiple Myeloma; MZL: Marginal Zone Lymphoma; PCL: Plasma Cell Leukemia; WM: Waldenström Macroglobulinemia.
Table 1: Case reports of patients with plasma cell dyscrasias who presented with hemoptysis as prominent symptom.
Conclusion
With this paper we describe bleeding diathesis as an unusual manifestation of IgG paraproteinemia in 2 patients that their primary symptom was hemoptysis. We propose therefore the addition of at least serum electrophoresis in the laboratory evaluation of hemoptysis and other coagulation disorders and bleeding symptoms in general. Moreover, the absence of coagulation abnormalities (and what is more, normal platelet count) in patients that present with recently developed bleeding diathesis and without a relevant personal or family history should direct the physician primarily towards their evaluation for AVWS and an underlying disease.
References
- McKenna RW, Kyle RA, Kuehl WM, Harris NL, Coupland RW, Fend F. Plasma cell neoplasms. Editors. In: Swerdlow S, Campo E, Lee Harris N, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue (IARC WHO Classification of Tumours) 4th Edition. The International Agency for Research on Cancer, Lyon. 2008: 200-213.
- Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014; 15: e538-e548.
- Schmidt T, Callander N. Diagnosis and Management of Monoclonal Gammopathy and Smoldering Multiple Myeloma. J Natl Compr Canc Netw. 2020; 18: 1720-1729.
- Merlini G, Palladini G. Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology Am Soc Hematol Educ Program. 2012; 2012: 595-603.
- Berger F, Traverse-Glehen A, Felman P, Callet-Bauchu E, Baseggio L, Gazzo S, et al. Clinicopathologic features of Waldenstrom’s macroglobulinemia and marginal zone lymphoma: are they distinct or the same entity? Clin Lymphoma. 2005; 5: 220-224.
- Zangari M, Elice F, Fink L, Tricot G. Hemostatic dysfunction in paraproteinemias and amyloidosis. Semin Thromb Hemost. 2007; 33: 339- 349.
- Coppola A, Tufano A, Di Capua M, Franchini M. Bleeding and thrombosis in multiple myeloma and related plasma cell disorders. Semin Thromb Hemost. 2011; 37: 929-945.
- Fermand JP, Bridoux F, Dispenzieri A, Jaccard A, Kyle RA, Leung N, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. 2018; 132: 1478-1485.
- Eby CS. Bleeding and thrombosis risks in plasma cell dyscrasias. Hematology Am Soc Hematol Educ Program. 2007: 158-164.
- Eby C. Pathogenesis and management of bleeding and thrombosis in plasma cell dyscrasias. Br J Haematol. 2009; 145: 151-163.
- Richard C, Cuadrado MA, Prieto M, Batlle J, López Fernández MF, Rodriguez Salazar ML, et al. Acquired von Willebrand disease in multiple myeloma secondary to absorption of von Willebrand factor by plasma cells. Am J Hematol. 1990; 35: 114-117.
- Mohri H, Tanabe J, Ohtsuka M, Yoshida M, Motomura S, Nishida S, et al. Acquired von Willebrand disease associated with multiple myeloma; characterization of an inhibitor to von Willebrand factor. Blood Coagul Fibrinolysis. 1995; 6: 561-566.
- Auwerda JJ, Sonneveld P, Leebeek FW. Temporary relief of symptomatic Von Willebrand disease by multiple myeloma. J Thromb Haemost. 2005; 3: 1088-1089.
- Djunic I, Elezovic I, Ilic V, Tomin D, Suvajdzic-Vukovic N, Bila J, et al. Acquired von Willebrand syndrome in multiple myeloma. Hematology. 2011; 16: 209-212.
- Jin N, Salahuddin FF, Nesbitt JA. Acquired von Willebrand disease and multiple myeloma: a case report of a breast cancer survivor. Blood Coagul Fibrinolysis. 2014; 25: 890-893.
- Dicke C, Schneppenheim S, Holstein K, Spath B, Bokemeyer C, Dittmer R, et al. Distinct mechanisms account for acquired von Willebrand syndrome in plasma cell dyscrasias. Ann Hematol. 2016; 95: 945-957.
- Katagiri S, Akahane D, Amano K, Ohyashiki K. Long-term remission of acquired von Willebrand syndrome associated with multiple myeloma using bortezomib and dexamethasone therapy. Haemophilia. 2016; 22: e557-e559.
- RicoRios N, Bowles L, Ayling RM. Multiple myeloma and acquired von Willebrand disease: a combined cause of preanalytical interference causing gel formation? Ann Clin Biochem. 2018; 55: 181-184.
- Kawashima I, Takano K, Kumagai T, Koshiishi M, Oishi S, Sueki Y, et al. Combined Coagulopathy Can Induce Both Hemorrhagic and Thrombotic Complications in Multiple Myeloma. Intern Med. 2018; 57: 3303-3306.
- Hinterleitner C, Pecher AC, Kreißelmeier KP, Budde U, Kanz L, Kopp HG, et al. Disease progression and defects in primary hemostasis as major cause of bleeding in multiple myeloma. Eur J Haematol. 2020; 104: 26-35.
- Franchini M, Gandini G, Di Paolantonio T, Mariani G. Acquired hemophilia A: a concise review. Am J Hematol. 2005; 80: 55-63.
- Muzaffar J, Katragadda L, Haider S, Javed A, Anaissie E, Usmani S. Rituximab and intravenous immunoglobulin (IVIG) for the management of acquired factor VIII inhibitor in multiple myeloma: case report and review of literature. Int J Hematol. 2012; 95: 102-106.
- Brás GP, Pinto RJ, Carvalho MM, Fernandes SP, Andrade JJ, Guimarães JE. Bortezomib: Potential Key Role in the Treatment of Multiple Myeloma- Related Acquired Hemophilia A. Semin Thromb Hemost. 2017; 43: 109-112.
- Pinchover LB, Alsharif R, Bernal T. Acquired haemophilia a secondary to multiple myeloma: management of a patient with a mechanical mitral valve. BMJ Case Rep. 2020; 13: e230798.
- Jalowiec KA, Andres M, Taleghani BM, Musa A, Dickenmann M, Angelillo- Scherrer A, et al. Acquired hemophilia A and plasma cell neoplasms: a case report and review of the literature. J Med Case Rep. 2020; 14: 206.
- Lackner H, Hunt V, Zucker MB, Pearson J. Abnormal fibrin ultrastructure, polymerization, and clot retraction in multiple myeloma. Br J Haematol. 1970; 18: 625-636.
- Carr ME Jr, Zekert SL. Abnormal clot retraction, altered fibrin structure, and normal platelet function in multiple myeloma. Am J Physiol. 1994; 266: H1195-H1201.
- Saif MW, Allegra CJ, Greenberg B. Bleeding diathesis in multiple myeloma. J Hematother Stem Cell Res. 2001; 10: 657-660.
- Kotlín R, Sobotková A, Riedel T, Salaj P, Suttnar J, Reicheltová Z, et al. Acquired dysfibrinogenemia secondary to multiple myeloma. Acta Haematol. 2008; 120: 75-81.
- Dear A, Brennan SO, Sheat MJ, Faed JM, George PM. Acquired dysfibrinogenemia caused by monoclonal production of immunoglobulin lambda light chain. Haematologica. 2007; 92: e111-e117.
- Post GR, James L, Alapat D, Guillory V, Cottler-Fox M, Nakagawa M. A case of acquired dysfibrinogenemia in multiple myeloma treated with therapeutic plasma exchange. Transfus Apher Sci. 2013; 48: 35-38.
- Huang H, Li H, Li D. Effect of serum monoclonal protein concentration on haemostasis in patients with multiple myeloma. Blood Coagul Fibrinolysis. 2015; 26: 555-559.
- Siddiq N, Bergstrom C, Anderson LD Jr, Nagalla S. Bleeding due to acquired dysfibrinogenemia as the initial presentation of multiple myeloma. BMJ Case Rep. 2019; 12: e229312.
- Spicka I, Rihova Z, Kvasnicka J, Cieslar P, Prochazka B, Klener P. Disturbances of anticoagulation and fibrinolytic systems in monoclonal gammopathies-another mechanism of M-protein interference with hemostasis. Thromb Res. 2003; 112: 297-300.
- Cesarman-Maus G, Braggio E, Maldonado H, Fonseca R. Absence of tissue factor expression by neoplastic plasma cells in multiple myeloma. Leukemia. 2012; 26: 1671-1674.
- Sugai S. IgA pyroglobulin, hyperviscosity syndrome and coagulation abnormality in a patient with multiple myeloma. Blood. 1972; 39: 224-237.
- Gabriel DA, Smith LA, Folds JD, Davis L, Cancelosi SE. The influence of immunoglobulin (IgG) on the assembly of fibrin gels. J Lab Clin Med. 1983; 101: 545-552.
- O’Kane MJ, Wisdom GB, Desai ZR, Archbold GP. Inhibition of fibrin monomer polymerization by myeloma immunoglobulin. J Clin Pathol. 1994; 47: 266-268.
- Panzer S, Thaler E. An acquired cryoglobulinemia which inhibits fibrin polymerization in a patient with IgG kappa myeloma. Haemostasis. 1993; 23: 69-76.
- De Maertelaere E, Castoldi E, Van Haute I, Deeren D, Devreese KM. The interaction of factor V and tissue factor pathway inhibitor in a myeloma patient with acquired factor V deficiency. Haemophilia. 2018; 24: e160-e163.
- Jia J, Wang H, Wu M, Zhang F, Liu X, Chen W, et al. Factor X Deficiency Caused by Nonsecretory Myeloma Successfully Corrected with Bortezomib: A Case Report and Review of the Literature. Acta Haematol. 2018; 140: 46-50.
- Reynolds SB, Maghavani DP, Hashmi H. Acquired factor X deficiency in a patient with multiple myeloma: a rare case highlighting the significance of comprehensive evaluation and the need for antimyeloma therapy for bleeding diathesis. BMJ Case Rep. 2019; 12: e230249.
- Torjemane L, Guermazi S, Ladeb S, Ben Romdhane N, Lakhal A, Abdelkefi A, et al. Heparin-like anticoagulant associated with multiple myeloma and neutralized with protamine sulfate. Blood Coagul Fibrinolysis. 2007; 18: 279- 281.
- Willner CA, Chisti MM. Treatment of Bleeding Diathesis Associated with a Heparin-Like Anticoagulant in Plasma Cell Neoplasia Using Protamine. Case Rep Hematol. 2018; 2018: 4342301.
- Xiao X, Zhong HY, Zhang GS, Deng MY. Thrombotic thrombocytopenic purpura as initial and major presentation of multiple myeloma. J Thromb Thrombolysis. 2013; 36: 422-423.
- Cheah CY, Orlowski RZ, Manasanch EE, Oo TH. Thrombotic thrombocytopenic purpura in a patient with lenalidomide-responsive multiple myeloma. Ann Hematol. 2015; 94: 1605-1607.
- Wallace MR, Simon SR, Ershler WB, Burns SL. Hemorrhagic diathesis in multiple myeloma. Acta Haematol. 1984; 72: 340-342.
- Friederich PW, Wever PC, Briët E, Doorenbos CJ, Levi M. Successful treatment with recombinant factor VIIa of therapy-resistant severe bleeding in a patient with acquired von Willebrand disease. Am J Hematol. 2001; 66: 292-294.
- Hayashi T, Yagi H, Suzuki H, Nonaka Y, Nomura T, Sakurai Y, et al. Lowdosage intravenous immunoglobulin in the management of a patient with acquired von Willebrand syndrome associated with monoclonal gammopathy of undetermined significance. Pathophysiol Haemost Thromb. 2002; 32: 33- 39.
- Krebs M, Meyer B, Quehenberger P, Turecek PL, Hejna M, Sperr WR, et al. Massive postoperative intramuscular bleeding in acquired von Willebrand’s disease. Ann Hematol. 2002; 81: 394-396.
- Huang YW, Saidi P. Acquired von Willebrand disease in a patient with monoclonal gammopathy of undetermined significance. Am J Med Sci. 2004; 327: 98-101.
- Maddox JM, Anderson JA, Plews D, Ludlam CA. Management of acquired von Willebrand’s syndrome in a patient requiring major surgery. Haemophilia. 2005; 11: 633-637.
- Michiels JJ, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, Budde U, et al. Immune-mediated etiology of acquired von Willebrand syndrome in systemic lupus erythematosus and in benign monoclonal gammopathy: therapeutic implications. Semin Thromb Hemost. 2006; 32: 577-588.
- Eikenboom JC, Tjernberg P, Van Marion V, Heering KJ. Acquired von Willebrand syndrome: diagnostic problems and therapeutic options. Am J Hematol. 2007; 82: 55-58.
- Karger R, Weippert-Kretschmer M, Budde U, Kretschmer V. Diagnosis and therapeutic management in a patient with type 2B-like acquired von Willebrand syndrome. Blood Coagul Fibrinolysis. 2011; 22: 144-147.
- Voisin S, Hamidou M, Lefrançois A, Sigaud M, Mahé B, Trossaërt M. Acquired von Willebrand syndrome associated with monoclonal gammopathy: a single-center study of 36 patients. Medicine (Baltimore). 2011; 90: 404-411.
- Puronen CE, Josephson NC, Broudy VC. Acquired von Willebrand syndrome in a patient with monoclonal gammopathy of undetermined significance. Blood Coagul Fibrinolysis. 2013; 24: 430-432.
- Al-Jafar H, Al-Barjas H, Hashem RA, Refaii TM, AlSaeed AM. Successful management of severe refractory acquired immune bleeding disorder: Prior to insisting surgery. Int J Surg Case Rep. 2014; 5: 1186-1189.
- Howard CR, Lin TL, Cunningham MT, Lipe BC. IgG kappa monoclonal gammopathy of undetermined significance presenting as acquired type III Von Willebrand syndrome. Blood Coagul Fibrinolysis. 2014; 25: 631-633.
- Yujiri T, Nakamura Y, Oota I, Nakano K, Tanizawa Y. Acquired von Willebrand syndrome associated with monoclonal gammopathy of undetermined significance. Ann Hematol. 2014; 93: 1427-1428.
- Lavin M, Brophy TM, Rawley O, O’Sullivan JM, Hayden PJ, Browne PV, et al. Lenalidomide as a novel treatment for refractory acquired von Willebrand syndrome associated with monoclonal gammopathy. J Thromb Haemost. 2016; 14: 1200-1205.
- Thomas W, Warner E, Cameron E, Symington E. Successful treatment of acquired von Willebrand disease with lenalidomide leading to dramatic resolution of intractable gastrointestinal bleeding: A case report. Haemophilia. 2018; 24: e140-e142.
- Désage S, Le Quellec S, Karlin L, Lienhart A, Meunier S, Rugeri L. A Prolonged Treatment Response in Acquired Von Willebrand Syndrome. Hamostaseologie. 2019; 39: 409-412.
- Kagaya M, Takahashi H. A case of type I cryoglobulinemia associated with a monoclonal gammopathy of undetermined significance (MGUS). J Dermatol. 2005; 32: 128-131.
- Duarte JM, Ocampo P, Ramos SG, Carballo OG, Barcia RE, Arévalo CE. Type-I Cryoglobulinaemia Associated to Monoclonal Gammapathy of Undetermined Significance. Prague Med Rep. 2020; 121: 194-199.
- Rassy EE, Tabchi S, Haddad FG, Chebib R, Assi T, Kourie HR, et al. Acquired factor inhibitor in a patient with monoclonal gammopathy of undetermined significance responding to rituximab. Blood Coagul Fibrinolysis. 2016; 27: 822-824.
- Rossi D, De Paoli L, Franceschetti S, Capello D, Vendramin C, Lunghi M, et al. Prevalence and clinical characteristics of immune thrombocytopenic purpura in a cohort of monoclonal gammopathy of uncertain significance. Br J Haematol. 2007; 138: 249-252.
- Boissier E, Darnige L, Dougados J, Arlet JB, Dupeux S, Georgin-Lavialle S, et al. Syndrome de Willebrand acquis: étude d’une série de neuf patients et revue de la littérature [Acquired von Willebrand syndrome: a case series of nine patients and literature review]. Rev Med Interne. 2014; 35: 154-159.
- Tefferi A, Hanson CA, Kurtin PJ, Katzmann JA, Dalton RJ, Nichols WL. Acquired von Willebrand’s disease due to aberrant expression of platelet glycoprotein Ib by marginal zone lymphoma cells. Br J Haematol. 1997; 96: 850-853.
- Komeno Y, Shibuya N, Uryu H, Yamada H, Toda T, Shibasaki M, et al. Splenic Marginal Zone Lymphoma with Acquired von Willebrand Syndrome Diagnosed via Splenic Bleeding. Intern Med. 2017; 56: 557-562.
- Meenhuis A, van Vliet R, Hudig F, Ypma PF, Schipperus MR, Hollestelle MJ. Successful treatment of a noninhibitory antibody-mediated acquired factor X deficiency in a patient with marginal-zone lymphoma. Clin Case Rep. 2015; 3: 587-593.
- Gicart M, De Weweire A, Demulder A, De Wilde V. Acquired hemophilia A revealed by spontaneous bleeding in an 80-year-old man: a marginal diagnosis? Acta Clin Belg. 2020: 1-3.
- Bouma B, Maas C, Hazenberg BP, Lokhorst HM, Gebbink MF. Increased plasmin-alpha2-antiplasmin levels indicate activation of the fibrinolytic system in systemic amyloidoses. J Thromb Haemost. 2007; 5: 1139-1142.
- Vaxman I, Gertz M. Recent Advances in the Diagnosis, Risk Stratification, and Management of Systemic Light-Chain Amyloidosis. Acta Haematol. 2019; 141: 93-106.
- Palumbo A, Rajkumar SV, Dimopoulos MA, Richardson PG, San Miguel J, Barlogie B, et al; International Myeloma Working Group. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia. 2008; 22: 414-423.
- Riccardi A, Gobbi PG, Ucci G, Bertoloni D, Luoni R, Rutigliano L, et al. Changing clinical presentation of multiple myeloma. Eur J Cancer. 1991; 27: 1401-1405.
- Turhal N, Henehan MD, Kaplan KL. Multiple myeloma: a patient with unusual features including intracranial and meningeal involvement, testicular involvement, organomegaly, and plasma cell leukemia. Am J Hematol. 1998; 57: 51-56.
- Kyle RA, Gertz MA, Witzig TE, Lust JA, Lacy MQ, Dispenzieri A, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003; 78: 21-33.
- Koduri PR, Gowrishankar S, Malladi VK. Variant morphology in multiple myeloma. Indian J Hematol Blood Transfus. 2014; 30: 86-87.
- Huang H, Bazerbachi F, Mesa H, Gupta P. Asymptomatic Multiple Myeloma Presenting as a Nodular Hepatic Lesion: A Case Report and Review of the Literature. Ochsner J. 2015; 15: 457-467.
- Thomas AS, Mehta A, Hughes DA. Gaucher disease: haematological presentations and complications. Br J Haematol. 2014; 165: 427-440.
- Zimran A, Ruchlemer R, Revel-Vilk S. A patient with Gaucher disease and plasma cell dyscrasia: bidirectional impact. Hematology Am Soc Hematol Educ Program. 2020; 2020: 389-394.
- Mendes FR, Sobral KM, Culler HF, Couto SCF, Pereira J, Rocha V, et al. Acquired hemophagocytic lymphohistiocytosis as initial manifestation of multiple myeloma: A case report and literature review. Medicine (Baltimore). 2020; 99: e22299.
- Cook J, Muchtar E, Warsame R. Updates in the Diagnosis and Management of AL Amyloidosis. Curr Hematol Malig Rep. 2020; 15: 155-167.
- Dispenzieri A. POEMS syndrome: 2017 Update on diagnosis, risk stratification, and management. Am J Hematol. 2017; 92: 814-829.
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003; 121: 749-757.
- Vetto JT, Beer TM, Fidda N, Ham B, Jimenez-Lee R, Schmidt W. Fineneedle aspiration diagnosis of plasmacytoma presenting as breast masses in a patient on estrogen therapy for prostate cancer. Diagn Cytopathol. 2004; 31: 417-419.
- Brady-West DC, Buchner-Daley LM, McGrowder DA, Taylor-Houston J, West KA. Multiple myeloma presenting as cervical lymphadenopathy in a 16-year-old boy. J Natl Med Assoc. 2009; 101: 810-812.
- Li K, Ku G, Yan M, Broome HE, Kelner M, Wang HY. A rare and unique case of aggressive IgE-λ plasma cell myeloma in a 28-year-old woman presented initially as an orbital mass. Hum Pathol. 2012; 43: 2376-2384.
- Abrar S, Ali N, Qureshi BM, Abbasi AN. Extramedullary plasmacytoma: rare neoplasm of parotid gland. BMJ Case Rep. 2018; 2018: bcr2017222367.
- Naymagon L, Abdul-Hay M. Primary extramedullary plasmacytoma with diffuse lymph node involvement: a case report and review of the literature. J Med Case Rep. 2019; 13: 153.
- Bíró L, Domján G, Falus A, Jakab L, Cseh K, Kalabay L, et al. Cytokine regulation of the acute-phase protein levels in multiple myeloma. Eur J Clin Invest. 1998; 28: 679-686.
- Alexandrakis MG, Passam FH, Ganotakis ES, Sfiridaki K, Xilouri I, Perisinakis K, et al. The clinical and prognostic significance of erythrocyte sedimentation rate (ESR), serum interleukin-6 (IL-6) and acute phase protein levels in multiple myeloma. Clin Lab Haematol. 2003; 25: 41-46.
- Niléhn JE, Nilsson IM. Coagulation studies in different types of myeloma. Acta Med Scand Suppl. 1966; 445: 194-199.
- Perkins HA, MacKenzie MR, Fudenberg HH. Hemostatic defects in dysproteinemias. Blood. 1970; 35: 695-707.
- Penny R, Castaldi PA, Whitsed HM. Inflammation and haemostasis in paraproteinaemias. Br J Haematol. 1971; 20: 35-44.
- Lackner H. Hemostatic abnormalities associated with dysproteinemias. Semin Hematol. 1973; 10: 125-133.
- Delanghe J, Langlois M, De Buyzere M. Congenital anhaptoglobinemia versus acquired hypohaptoglobinemia. Blood. 1998; 91: 3524.
- Laffan MA, Lester W, O’Donnell JS, Will A, Tait RC, Goodeve A, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol. 2014; 167: 453- 465.
- Bolaman Z, Yavasoglu I, Unubol M, Kadikoylu G. Hemoptysis as a presenting sign of multiple myeloma. Respir Med. 2008; 102: 1672-1673.
- Morita Y, Yasuda M, Nakao M, Tsujimura Y, Isono M. Pulmonary hemorrhage and acute renal failure as an initial presentation of multiple myeloma. Intern Med. 2010; 49: 1401-1403.
- Szeto JS, Perez JA Jr. Multiple myeloma presenting as pulmonary renal syndrome. Methodist Debakey Cardiovasc J. 2013; 9: 58-59.
- Coucke L, Marcelis L, Deeren D, Van Dorpe J, Lambein K, Devreese K. Lymphoplasmacytic lymphoma exposed by haemoptysis and acquired von Willebrand syndrome. Blood Coagul Fibrinolysis. 2014; 25: 395-397.
- Clay RD, Utz JP. Coital Hemoptysis Due to Cardiac AL Amyloidosis. Mayo Clin Proc. 2018; 93: 811-812.
- Uwingabiye J, Zahid H, El Amrani M, Labrini F, Elkhazraji A, El Kabbaj D, et al. Rare and unusual case of anti-factor XI antibodies in patient with plasma cell leukemia. BMC Hematol. 2018; 18: 18.