
Case Report
Ann Hematol Oncol. 2021; 8(3): 1334.
IgG4-Related Disease and Successful Treatment with Rituximab in a Three-Year-Old Boy
Marissen J1*, Pagel J2, Steinmetz A2, Härtel C1,2 and Melchior Lauten2
¹Department of Pediatrics, University of Wuerzburg, Germany
²Department of Pediatrics and Adolescent Medicine, University of Luebeck, Germany
*Corresponding author: Marissen J, Department of Pediatrics, University of Wuerzburg, University Hospital Wuerzburg, Josef-Schneider-Straße 2, 97080 Würzburg, Germany
Received: February 04, 2021; Accepted: March 09, 2021; Published: March 16, 2021
Abstract
IgG4-Related Disease (IgG4-RD) is a rare inflammatory disease, which may affect all organs and lead to severe fibrosis and organ damage. Mainly described in Japanese or Asian adults, case reports on children-variously affected by the disease-are increasing worldwide. While glucocorticoids are broadly used as first line treatment, several cases require second line options due to relapse or resistance to first line treatment.
We report a boy who was diagnosed with IgG4-related disease at the age of three years. He presented an IgG4-associated tumorous lesion in the upper lobe of the left lung, pleural and pericardial effusion and enlarged abdominal lymph nodes. Therapy with surgery and glucocorticoids followed by rituximab treatment led to continuous complete remission 27 months after diagnosis of IgG4-RD.
Keywords: IgG4 related disease; IgG4; Lung tumor; RituximabAbbreviations
IgG: Immunoglobulin G; IgG4-RD: Immunoglobulin G4 Related Disease
Case Presentation
The boy was initially admitted to an Armenian hospital at the age of three years due to pneumonia and high fever. Pulmonary imaging showed lesions of the mediastinum and the upper lobe of the left lung. Lymph node biopsy was suspicious for a mediastinal large B-cell lymphoma. As the patient was clinically well at that time, the parents did not agree on further biopsies. Due to progression of the tumor six months later, a second biopsy was performed, again implicating a large B-cell lymphoma. However, reference pathology favored the diagnosis of benign and reactive lymph node disease.
The patient was then admitted to our hospital. On admission, he presented without fever or loss of weight, however, he showed significant night sweats. Magnetic resonance imaging revealed a centrally calcified tumor in the left mediastinum without metastases (Figure 1). Thoracoscopy confirmed a tumor in the upper lobe of the left lung. Laboratory tests initially showed mild anemia (9g/ dl), thrombocytosis (654x109/l), and normal white blood count with lymphocytosis (63.9%). Immune status revealed elevated CD4 (2.43x109/l) and CD8 (1.920x109/l) positive lymphocytes. Thyroid parameters, liver and renal parameters were within normal ranges and no elevation of inflammation parameters was found at that time. Alpha fetoprotein, lactate dehydrogenase and human chorionic gonadotropin were normal and infectious disease screening (including Epstein-Barr-Virus, multiplex PCR for viruses and QuantiFERON-test) was unremarkable. Antinuclear antibodies were within normal ranges.
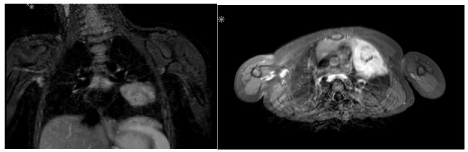
Figure 1: Magnetic resonance imaging of the tumorous lesion.
Due to the growing tumor with unknown origin surgical resection of the left upper lobe of the lung was performed. Histopathology revealed a plasma-cellular tumorous lesion measuring 5.8cm in diameter and showing fibrosis. IgG4 was found to be positive in the tumor (95 IgG4 cells per high powered field, IgG4/IgG 38%). As also the IgG4 serum level was elevated (IgG 16g/l (4.2-9.2 g/L), IgG1 11.1g/l (2.7-8.1 g/l), IgG2 4.08g/l (0.65-2.2 g/l), IgG3 0.96g/l (0.16- 0.96 g/l), IgG4 2.11g/l (0.012-0.938 g/l), IgA 2.03g/l (0.3-1.9 g/l), IgM 1.25g/l (0.52-1.9 g/l)), we diagnosed IgG4-related disease (IgG4-RD). Staging with ultrasound revealed enlarged abdominal lymph nodes but no further involvement of solid organs.
Oral prednisolone (1mg/kg/day) was initiated and the patient was soon discharged from hospital in good clinical condition.
Ultrasound ten days after discharge revealed pleural and pericardial effusion not necessitating drainage. In addition, cholecystitis was suspected by ultrasonography. The boy was readmitted to the hospital. Due to these findings, we decided to initiate rituximab (anti-CD-20 antibody), which was administered with four doses (375mg/m2) in weekly intervals.
Within two months, serum IgG4 levels normalized (Figure 2). Ten weeks after initiation of glucocorticoids and two weeks after the fourth rituximab treatment we tapered prednisolone to a daily dose below the adjusted Cushing threshold dose (6mg/m²). Following rituximab, B-cell levels were low (lowest level: <0.001x109/l 28 days after the fourth dose) and immunoglobulin levels fell slightly (minimum IgG 6.09g/l three months after end of rituximab therapy). Prednisolone treatment was continued at 4mg per day for another five months, then further tapered and stopped 21 months after initiation of therapy. Cotrimoxazole was discontinued after nine months.
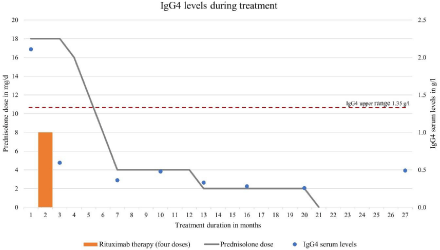
Figure 2: IgG4 serum levels during treatment.
The boy was followed up as an outpatient every six to twelve weeks. He was admitted to hospital once due to diarrhea, fever and otitis media and received antibiotic treatment with amoxicillin. During winter, the boy presented with recurrent infections of the upper respiratory tract, but no antibiotic or immunoglobulin treatment had to be initiated. B-cells normalized within ten months after the initiation of rituximab. The patient was seen regularly by ophthalmologists; however, no ocular affection or side effects of glucocorticoid treatment were seen. Magnetic resonance imaging was not suspicious for the development of tumorous lesions or organ fibrosis. To date, 27 months after start of therapy and 6 months after cessation of prednisolone, the boy shows no clinical or laboratory signs of IgG4-RD.
Discussion
We present a three-year-old boy with IgG4-RD successfully treated with corticosteroids, surgery and rituximab. IgG4-RD is a rare inflammatory disease which can affect nearly all organs, but mainly concerns the pancreas, biliary tract, glands, lymph nodes and the retro-peritoneum. The inflammation within the organs can lead to fibrosis and, consequently, to severe organ damage [1-3]. Literature reports and consensus statements on IgG4-RD mainly concentrate on adults, but children are also known to be affected [4]. However, there is no consensus for the diagnosis and treatment of IgG4- related disease in children. In the existing literature approximately 30 children with IgG4-RD have been reported [5-9]. In adults and in children, the affection of organs varies widely [4-15].
The diagnosis of IgG4-RD is established using clinical and laboratory parameters as well as biopsy and imaging. As most symptoms and findings of IgG4-RD have a broad differential diagnosis [16], a step-by-step approach is required [1]. Interestingly, the diagnosis of IgG4-RD does not necessarily require elevated IgG4 serum levels [3,6,17], but elevated IgG4 levels are associated with multiple organ involvement [2]. In line with that, IgG4 levels above 135 mg/dl, >40% of IgG4+ plasma cells (IgG4/IgG) and >10 cells/high powered field of biopsy sample are being proposed to proof IgG4- RD [18]. Our patient presented the above-mentioned criteria with the exception of the IgG4+ plasma cells, which were at 38%.
First line treatment using glucocorticoids is recommended to all symptomatic patients or patients with severe organ manifestation in order to avoid irreversible organ fibrosis [2,14]. In children, 0.5- 2 mg/kg/day prednisone is suggested followed by slow tapering [4]. Tapering of glucocorticoids in adults is recommended after weeks to months [1,2], even though studies described higher relapse rates in patients after early tapering or discontinuation [2]. Japanese guidelines recommend glucocorticoid maintenance for up to three years [19]. Still, the optimal duration of maintenance remains unclear [2].
In children requiring second line treatment for relapse, nonresponse or contraindication to glucocorticoids the use of azathioprine [7], mycophenolate mofetil [12], methotrexate [5], adalimumab, cyclophosphamide, colchicine [4] and surgical intervention [6,10] has been reported.
Rituximab is a monoclonal anti-CD20 antibody, which depletes circulating B-cells [20]. Regarding IgG4-RD, it has been used in small adult cohorts and proved to be effective for remission induction measured by normalization or improvement of IgG4 serum levels and successful glucocorticoid discontinuation [11,21-23]. We found reports about only four children receiving rituximab for the treatment of IgG4-RD [15,24-26]: A 12-year-old girl with IgG4-RD received rituximab due to severe refractory disease (two doses of 750mg/m2 every 14 days). She relapsed one year later, again received rituximab and achieved continuous complete remission [15]. Another 12-yearold girl received rituximab (two doses of 1000mg every 14 days) due to relapse while tapering prednisone [25]. Furthermore, a 15-year-old girl received rituximab 2x600 mg every 14 days due to insufficient response to prednisone treatment [26] and a 17-yearold boy with IgG4-related lymphadenopathy and refractory scleritis received two doses of rituximab (1000mg) after initial treatment with repeated courses of prednisone, methotrexate, cyclosporine and mycophenolate mofetil. After rituximab, he achieved continuous complete remission [24].
All reported cases had ophthalmic affection and presented with different organ involvement, but none showed pulmonary affection. No severe side effects of rituximab therapy were stated [15,24-26]. To our knowledge, our patient is the youngest patient reported receiving rituximab for IgG4-RD with pulmonary involvement. Generally, most children reported with IgG4-RD were older than ten years [4]. In the above-mentioned cases, rituximab was mainly used after months or years of glucocorticoid treatment. As our patient showed multiple progressive organ involvement despite prednisolone therapy, we decided for early rituximab therapy in order to prevent short and long-term glucocorticoid related toxicity. We did not observe significant acute toxic effects of rituximab therapy. B-cell depletion, however, is associated with an increased risk of infections, and long-term-follow-up including the renewal of the vaccination schedule is required.
In our patient, IgG4 levels normalized during treatment with glucocorticoids and rituximab within two months. Normalization of serum IgG4 levels is supposed to indicate disease inactivity during treatment [27] However, patients may relapse despite normal IgG4 serum levels [3,28], suggesting that IgG4 serum levels lack diagnostic specificity [3,5].
Conclusion
IgG4-RD is a rare disease presenting with a variety of phenotypes and affecting predominantly adults. Glucocorticoids are effective, but there is no consensus on second line treatment. Data on rituximab are scarce, especially in children. We report a case with continuous complete remission after surgery of the primary tumorous lesion, adjuvant treatment with glucocorticoids (1mg/kg/d) and four doses of rituximab (375mg/m2). We suggest rituximab to be considered as second-line treatment in children in order to prevent morbidity due to long-term glucocorticoid use. Prospective studies are required to better understand the disease and harmonize treatment of IgG4-RD in children.
References
- Yamamoto M, Takahashi H, Shinomura Y. Mechanisms and assessment of IgG4-related disease: lessons for the rheumatologist. Nat Rev Rheumatol. 2014; 10: 148-159.
- Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol. 2015; 67: 1688- 1699.
- Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012; 366: 539-551.
- Karim F, Loeffen J, Bramer W, Westenberg L, Verdijk R, van Hagen M, et al. IgG4-related disease: a systematic review of this unrecognized disease in pediatrics. Pediatr Rheumatol Online J. 2016; 14: 18.
- Tille L, Schnabel A, Laass MW, Hahn G, Taut H, Leszczynska A, et al. Orbital inflammation and colitis in pediatric IgG4-related disease: A case report and review of the literature. Eur J Rheumatol. 2019; 7: 1-7.
- Szczawinska-Poplonyk A, Wojsyk-Banaszak I, Jonczyk-Potoczna K, Breborowicz A. Pulmonary manifestation of immunoglobulin G4-related disease in a 7-year-old immunodeficient boy with Epstein-Barr virus infection: a case report. Ital J Pediatr. 2016; 42: 58.
- Bolia R, Chong SY, Coleman L, MacGregor D, Hardikar W, Oliver MR. Autoimmune Pancreatitis and IgG4 Related Disease in Three Children. ACG Case Rep J. 2016; 3: e115.
- Aydemir Y, Akcoren Z, Demir H, Saltik Temizel IN, Ozen H, Yuce A. Clinical and histopathological features of immunoglobulin G4-associated autoimmune hepatitis in children. J Gastroenterol Hepatol. 2019; 34: 742-746.
- Smerla RG, Rontogianni D, Fragoulis GE. Ocular manifestations of IgG4- related disease in children. More common than anticipated? Review of the literature and case report. Clin Rheumatol. 2018; 37: 1721-1727.
- Corujeira S, Ferraz C, Nunes T, Fonseca E, Vaz LG. Severe IgG4-Related Disease in a Young Child: A Diagnosis Challenge. Case Rep Pediatr. 2015; 2015: 140753.
- Pifferi M, Di Cicco M, Bush A, Caramella D, Chilosi M, Boner AL. Uncommon pulmonary presentation of IgG4-related disease in a 15-year-old boy. Chest 2013; 144: 669-671.
- Mannion M, Cron RQ. Successful treatment of pediatric IgG4 related systemic disease with mycophenolate mofetil: case report and a review of the pediatric autoimmune pancreatitis literature. Pediatr Rheumatol Online J. 2011; 9: 1.
- Ryu JH, Sekiguchi H, Yi ES. Pulmonary manifestations of immunoglobulin G4-related sclerosing disease. Eur Respir J. 2012; 39: 180-186.
- Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015; 385: 1460-1471.
- Gillispie MC, Thomas RD, Hennon TR. Successful treatment of IgG-4 related sclerosing disease with rituximab: a novel case report. Clin Exp Rheumatol. 2015; 33: 549-550.
- Ebbo M, Grados A, Bernit E, Vély F, Boucraut J, Harlé J-R, et al. Pathologies Associated with Serum IgG4 Elevation. Int J Rheumatol. 2012; 2012: 602809.
- Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2015; 74: 14-18.
- Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012; 22: 21-30.
- Kamisawa T, Okazaki K, Kawa S, Shimosegawa T, Tanaka M. Japanese consensus guidelines for management of autoimmune pancreatitis: III. Treatment and prognosis of AIP. J Gastroenterol. 2010; 45: 471-477.
- Randall KL. Rituximab in autoimmune diseases. Aust Prescr. 2016; 39: 131- 134.
- Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010; 62: 1755-1762.
- Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore). 2012; 91: 57-66.
- Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015; 74: 1171-1177.
- Caso F, Fiocco U, Costa L, Sfriso P, Punzi L, Doria A. Successful use of rituximab in a young patient with immunoglobulin G4-related disease and refractory scleritis. Joint Bone Spine. 2014; 81: 190-192.
- Sane M, Chelnis J, Kozielski R, Fasiuddin A. Immunoglobulin G4-related sclerosing disease with orbital inflammation in a 12-year-old girl. J AAPOS. 2013; 17: 548-550.
- Prabhu SM, Yadav V, Irodi A, Mani S, Varghese AM. IgG4-related disease with sinonasal involvement: A case series. Indian J Radiol Imaging. 2014; 24: 117-120.
- Tabata T, Kamisawa T, Takuma K, Egawa N, Setoguchi K, Tsuruta K, et al. Serial changes of elevated serum IgG4 levels in IgG4-related systemic disease. Intern Med. 2011; 50: 69-75.
- Kamisawa T, Shimosegawa T, Okazaki K, Nishino T, Watanabe H, Kanno A, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009; 58: 1504-1507.