
Case Report
Ann Hematol Oncol. 2021; 8(3): 1335.
EBV-Associated Post-Transplant Lymphoproliferative Disease (PTLD) in Allogeneic Transplantation
Cutini I¹*, Peruzzi B², Caporale R², Nozzoli C¹, Gozzini A¹, Innocenti C¹, Boncompagni R¹, Fani A¹ and Saccardi R¹
¹Department of Cellular Therapies and Transfusion Medicine, Careggi University Hospital, Italy
²Flow Cytometry Diagnostic Center and Immunotherapy (CDCI), Careggi University Hospital, Italy
*Corresponding author: Ilaria Cutini, Department of Cellular Therapies and Transfusion Medicine, Careggi University Hospital, Florence, Italy
Received: February 13, 2021; Accepted: March 09, 2021; Published: March 16, 2021
Abstract
Post-Transplant Lymphoproliferative Disease (PTLD) following both Solid Organ Transplantation (SOT) and Hematopoietic Stem Cell Transplantation (HSCT) is a rare life-threatening complication. The majority of PLTDs are associated to Epstein Bar Virus (EBV) [1] reactivation, usually in the early phase [2] after transplant, when the patient is severely immunocompromised and is unable to control virus replication [3]. Despite the mortality of EBV-associated PTLD has been reduced over the years, the different histological patterns of its presentation, ranging from indolent to high grade B cell lymphoma, still play a role in the outcome. Herein, we report the case of a 60-years-old man diagnosed with acute myeloid leukemia who underwent allogeneic transplantation and developed a fatal Hemophagocytic Histiocytosis (HLH) secondary to an aggressive EBV-associated PTLD, not responding to a rituximab-based treatment.
Keywords: EBV; PTLD; Allogeneic transplantation; HLH
Introduction
Epstein-Barr Virus (EBV) is an herpes virus affecting over 90% of the adult population worldwide [4], and it is responsible of severe HSCT complications such as PTLD and HLH. EBV virus infects B-cell lymphocytes and remains in an asymptomatic latent state. Intense immunosuppression, such as after HSCT, may result in viral reactivation, possibly leading to an EBV-related lymphoma, where an impaired T-lymphocyte immunity allows EBV to extend the lifespan of the infected B-lymphocytes and consequently increases their likelihood of developing mutations [5]. The occurrence of EBV-PTLD after HSCT ranges from 0.5% to 17% [6] and between 1% to 15% in recipients of SOTs [7]. Several risk factors have been described in HSCT most frequent being: reduced intensity Conditioning Regimen (RIC), donor type (unrelated fully or mismatched donor, sibling mismatched donor), EBV donor/recipient serology mismatched (negative recipient and positive donor), graft versus host disease (GVHD) prophylaxis (in vivo or ex vivo T cell depletion, Anti- Thymocyte Globulin (ATG), alemtuzumab), Peripheral Blood Stem Cells Source (PBSC) [8]. In SOTs the main risk factor is EBV donor/ recipient serology mismatched. Positive donor and negative recipient represent the highest risk combination for PTLD.
EBV-PTLD can be classified as probable or proven Probable EBV disease: significant lymphadenopathy, hepatosplenomegaly or other end-organ manifestations (without tissue biopsy, but in the absence of other documented cause), together with significant EBV DNAemia. Proven EBV disease: detection of EBV nucleic acids or EBVencoded proteins in a tissue specimen, together with symptoms and/ or signs from the affected organ [9].
Therefore, blood monitoring of EBV-DNA is mandatory for HSCT recipient during the follow up to prevent PTLD.
The treatment of PTLD is based on: 1) the administration of rituximab; 2) reduction of immunosuppression; 3) EBV-cytotoxic T lymphocytes and donor lymphocyte infusion when no signs or symptoms of GVHD are detected and 4) chemotherapy for rituximabrefractory patients [9]. About 60% of patients respond after 1 or 2 rituximab infusions. Unfortunately, a rare evolution of EBV-PTLD in HSCT setting is HLH, which is associated to 80% mortality rate [10]. We report here a case of a massive EBV reactivation which quickly developed a fulminant HLH, two months after the transplantation.
Case Presentation
A 60-years-old man diagnosed with acute myeloid leukaemia ELN low risk (normal karyotype, NPM1 type A mutated, FLT3 ITD and D835 wild type) relapsed early after the consolidation therapy and underwent an allogeneic HSCT in second complete remission from a fully matched unrelated donor, following thiotepa-busulfanfludarabine myeloablative conditioning regimen in July 2020. GVHD prophylaxis consisted of cyclosporine, high dose of ATG plus a short course of methotrexate. Patient was IgG positive for EBV, CMV and Toxoplasmosis, whilst the donor was positive for EBV only. Bi-weekly monitoring of whole blood PCR for CMV, EBV, HHV6, adenovirus and toxoplasma were performed from the day of engraftment. The patient was discharge in good clinical condition with a fully recovered of the complete blood count. The bone marrow evaluations performed at 1 month after transplantation confirmed a complete haematological remission, negative minimal residual disease, negative NPM1 type A molecular analysis, and fully donor chimerism.
At +58 days from HSCT was admitted into hospital due to asthenia and light fever (37.2-37.6 °C). The blood cultures were negative. EBV PCR resulted positive and raised in 4 days from 2x103 to 3x106 viral genome/ml. A total body CT scan revealed previously unreported splenomegaly and multiple abdominal lymph nodes (diameter of 1,5 cm long). The bone marrow evaluation performed at +60 days from HSCT showed 20% of large lymphocytes with medium nuclear/cytoplasmic ratio, blue cytoplasm, many of them containing granules, and tight chromatin. No evidence of leukaemia recurrence was detected.
Multiparameter Flow Cytometry (MFC) performed on bone marrow blood (Figure 1, panel A in fuchsia) described a population of abnormal B lymphocytes co-expressing CD19+, CD20+, CD22+, CD79b+, CD38+, and clonal K+ light, and both forward and side scatters were high. No expression of CD5, CD23 and CD10 were detected. Other normal populations were identified such as B lymphocyte progenitors (Figure 1, Panel A in orange and blue) and polyclonal mature B-lymphocytes (Figure 1, Panel A in green). MFC was suggestive of B-cell lymphoma. In the peripheral blood smear, a population of 18% of large size lymphocytes was observed. MFC confirmed the same surface antigens expression of the blood marrow evaluation (Figure 1, panel B). All these evidences were indicative of an EBV-associated lymphoproliferative disease. Promptly, cyclosporine was withdrawn and Rituximab 375mg/sqm was administered with no clinical response, whilst EBV PCR assay reached 6x106 viral genome/ ml in 4 more days.
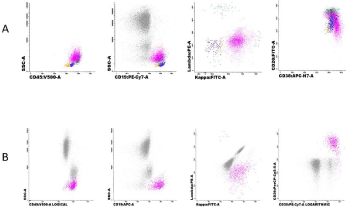
Figure 1:
On day 63 from HSCT seizure and lethargic state were shown: electroencephalography showed a diffuse cerebral dysfunction with alterations at brain MRI scan associated with high fever (TC 39°C); the patient was then transferred to intensive care unit and started invasive ventilation. Due to the patient’s critical conditions there was no time to perform cerebrospinal fluid analysis and Positron Emission Tomography (PET). A worsening of both haemoglobin (dropping down form 11,0 g/dl to 7,2g/dl) and platelet count (from 56x1012/L to 37x1012/L), the persistence of the fever (39.3°C), a quickly increase of ferritin level from 5800 to 31000 ng/ml (Normal Value (nv) 13441 ng/ml), serum glutamic oxaloacetic transaminase 56 U/L (nv <30 U/L), and increased LDH 1093 U/L (nv 135-225 U/L) were suggestive of secondary HLH as an end-stage feature of EBV-associated PTLD. A bone marrow smear was performed, several hemophagocytosis features were observed (Figure 2) with the persistence of clonal B lymphocytes not bearing CD20 surface antigen, likely due to Rituximab treatment. A diagnosis of HLH was carried out based on HS score [11-13] (218 points, resulting in a probability of 93-96 % of HLH). Dexamethasone 40mg daily was administered. The patient died 8 days after the admission, at +66 days after HSCT.
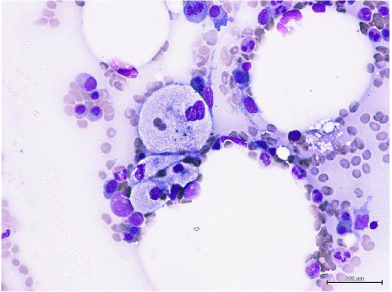
Figure 2:
Discussion
PTLD is a heterogeneous group of lymphoid disorders ranging from indolent to high grade lymphomas. The incidence of PTLD after HSCT varies from 1,2% in matched sibling donor to 4.0% in matched unrelated donor [9]. The median time to develop EBV-PTLD is 2-4 months after HSCT. Several risk factors for PTLD have been described, they can be distinguished in pre- (use of ATG or alemtuzumab, EBV serology donor/recipient mismatched, cord blood transplantation, HLA mismatched donor, splenectomy and second HSCT) and posttransplant (severe acute or chronic GVHD, high EBV viral load, treatment with mesenchymal stem cells). Our patient had 1 pre- transplant, (the inclusion of ATG in the conditioning regimen) and 1 post-transplant risk factors (high EBV viral load). According to the sixth European Conference of Infections in Leukemia (ECIL-6) guidelines [9] he belonged to high risk category due to the type of donor (matched unrelated donor). In agreement with Fujimoto A et al. [14] he had a probability of 4,6% at 2 years to develop PTLD.
PBSC source is not considered among the risk factors, however van Esser J et al. have shown its correlation with higher occurrence of PTLD. PBSC graft contains a higher number of EBV-infected B-cells lymphocytes and cytokines released by maturing myeloid progenitors which can stimulate the proliferation of EBV-infected B-cells [15].
The presence of splenomegaly is unusual, fever and lymphadenopathy are the most common presentation frequently associated with progressive multi-organ failure and death [16]. The diagnostic approach is based on biopsies of enlarged lymph nodes and/or on other sites of suspected EBV disease, however the clinical status of our patient did not allow to perform them, and a non-invasive strategy was preferred: EBVDNA-emia and total body CT scan [17]. Since neurological symptoms appeared, the patient conditions did not allow to assess whether a central nervous system EBV disease was present. MFC analysis in peripheral and marrow blood were suggestive of B-cell lymphoma resembling a Diffuse Large B Cell Lymphoma (DLBCL), which was consistent with the clinical presentation. That analysis was crucial to assessed the presence of a clonal B-cell lymphocytes and to carry out an EBV-PTLD diagnosis. The reduction of immunosuppression and rituximab administration are the standard treatment of choice with a response rate of 40-68 % [1], however, in our case it was ineffective and he rapidly developed HLH as end-organ disease. As we report, Wang et al. [19] and Yamamoto et al. [20] described a case of EBV-PTLD complicated with HLH in which despite multiple interventions the patients did not respond and died, whereas Weber et al. [18] reported an excellent outcome with the discontinuation of immunosuppression and the administration of dexamethasone and rituximab. Although the majority of infectionrelated secondary HLH respond to steroids and to the treatment of the primary cause, in this scenario the management of EBV-related HLH remains difficult [18] with a mortality rate of 80% [10].
A better understanding of the underlying mechanism involved in the development of HLH and EBV-PTLD are needed. Rituximab resistance confers an extremely poor prognosis in this peculiar setting. A balance between immunosuppression to control HLH and the withdrawn of the immunosuppression to control EBV remain the main challenge in this rare and mostly fatal post-transplant complication. That paper highlights the main unmet needs in clinical management and the lack of specific prognostic criteria, which might be useful in an early treatment intervention to preserve from the development of HLH.
Declaration
Authors contributions: IC, CN wrote the paper. AG, RB, AF and CI revised literature. BP and RC performed MFC analysis. RS revised the paper.
References
- Petrara MR, Giunco S, Serraino D, Dolcetti R, Rossi AD. Post-transplant lymphoproliferative disorders: From epidemiology to pathogenesis-driven treatment. Cancer Lett. 2015.
- Dolcetti R. B lymphocytes and Epstein-Barr virus: The lesson of posttransplant lymphoproliferative disorders. Autoimmun Rev. 2007.
- Rasche L, Kapp M, Einsele H, Mielke S. EBV-induced post transplant lymphoproliferative disorders: A persisting challenge in allogeneic hematopoetic SCT. Bone Marrow Transplant. 2014.
- Cohen JI. Epstein-Barr Virus Infection. N Engl J Med. 2000.
- Burns DM, Tierney R, Shannon-Lowe C, Croudace J, Inman C, Abbotts B, et al. Memory B-cell reconstitution following allogeneic hematopoietic stem cell transplantation is an EBV-associated transformation event. Blood. 2015.
- Curtis RE, Travis LB, Rowlings PA, Socie G, Kingma DW, Banks PM, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: A multi-institutional study. Blood. 1999; 94: 2208-2216.
- DeStefano CB, Desai SH, Shenoy AG, Catlett JP. Management of posttransplant lymphoproliferative disorders. Br J Haematol. 2018; 182: 330-343.
- Landgren O, Gilbert ES, Rizzo JD, Socie G, Banks PM, Sobocinski KA, et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood. 2009; 113: 4992-5001.
- Styczynski J, Van Der Velden W, Fox CP, et al. Management of epstein-barr virus infections and post-transplant lymphoproliferative disorders in patients after allogeneic hematopoietic stem cell transplantation: Sixth European Conference on Infections in Leukemia (ECIL-6) guidelines. Haematologica. 2016.
- Sandler RD, Carter S, Kaur H, Francis S, Tattersall RS, Snowden JA. Haemophagocytic Lymphohistiocytosis (HLH) following allogeneic Haematopoietic Stem Cell Transplantation (HSCT)-time to reappraise with modern diagnostic and treatment strategies? Bone Marrow Transplant. 2020; 55: 307-316.
- Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the hscore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014; 66: 2613-2620.
- Ghobrial IM, Habermann TM, Maurer MJ, Geyer SM, Ristow KM, Larson TS, et al. Prognostic analysis for survival in adult solid organ transplant recipients with post-transplantation lymphoproliferative disorders. J Clin Oncol. 2005.
- Bhatia S, Ramsay NKC, Steinbuch M, Dusenbery KE, Shapiro RS, Weisdorf DJ, et al. Malignant neoplasms following bone marrow transplantation. Blood. 1996.
- Fujimoto A, Hiramoto N, Yamasaki S, Inamoto Y, Uchida N, Maeda T, et al. Risk Factors and Predictive Scoring System For Post-Transplant Lymphoproliferative Disorder after Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2019; 25: 1441-1449.
- Van Esser JWJ, Van Der Holt B, Meijer E, Niesters HG, Trenschel R, Thijsen SF, et al. Epstein-Barr Virus (EBV) reactivation is a frequent event after allogeneic Stem Cell Transplantation (SCT) and quantitatively predicts EBVlymphoproliferative disease following T-cell-depleted SCT. Blood. 2001.
- Xuan L, Jiang X, Sun J, Zhang U, Huang F, Fan Z, et al. Spectrum of epsteinbarr virus-associated diseases in recipients of allogeneic hematopoietic stem cell transplantation. Transplantation. 2013; 96: 560-566.
- Fox CP, Burns D, Parker AN, Peggs KS, Harvey CM, Natarajan S, et al. EBV-associated post-transplant lymphoproliferative disorder following in vivo T-cell-depleted allogeneic transplantation: Clinical features, viral load correlates and prognostic factors in the rituximab era. Bone Marrow Transplant. 2014; 49: 280-286.
- Weber T, Wickenhauser C, Monecke A, Glaser C, Stadler M, Desole M, et al. Treatment of rare co-occurrence of Epstein-Barr virus-driven post-transplant lymphoproliferative disorder and hemophagocytic lymphohistiocytosis after allogeneic stem cell transplantation. Transpl Infect Dis. 2014.
- Wang IJ, Lu MY, Chiang BL, Lin WC, Lin DT, Lin KH, et al. Epstein-Barr virus associated post-transplantation lymphoproliferative disorder with hemophagocytosis in a child with Wiskott-Aldrich syndrome. Pediatr Blood Cancer. 2005; 45: 340-343.
- Yamamoto N, Nishimura N, Takeuchi M, Ito T, Yokozaki H, Hirase S, et al. The emergence of CD20-/CD19- tumor cells after rituximab therapy for Epstein–Barr virus-associated post-transplant lymphoproliferative disorder complicated with hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2014; 173: 1615-1618.