
Rapid Communication
Ann Hematol Oncol. 2021; 8(4): 1340.
Intermittent Transfusions for Treatment of Thalassemia in the State of Georgia, 2007-2016
Rollins MR1,2*, Boudreaux J2, Eckman J3, Branscomb J4, Zhou M4 and Snyder A4
1Department of Pathology, Emory University, USA
2Department of Pediatrics, Emory University, USA
3Department of Hematology & Oncology, Emory University, USA
4Georgia State University, Andrew Young School of Policy Studies, USA
*Corresponding author: Margo R Rollins, Department of Pathology, Emory University, School of Medicine, Center for Transfusion and Cellular Therapies, Emory University, 1001 Johnson Ferry RD, NE, Atlanta, Georgia 30342, USA
Received: March 04, 2021; Accepted: April 03, 2021; Published: April 10, 2021
Abstract
Background: Individuals with Non-Transfusion Dependent Thalassemia (NTDT) may require infrequent transfusions. Knowing transfusion history, while important, can be challenging in this subgroup.
Study Design: Hospital discharge data in Georgia (2007-2016) was reviewed. Thalassemia patients were defined as ≥3 encounters with a thalassemia diagnosis code. Transfusion was defined by the presence of a diagnosis, CPT, revenue, or HCPCS code for red cell transfusion.
Results: There were 428 patients identified; 57 received multi-site transfusions.
Conclusion: Georgia hospitals provide intermittent transfusions to low volumes of probable NTDT patients. Patient and provider education may help assure adherence to best practices, avoiding serious transfusion complications.
Keywords: Non-transfusion dependent thalassemia; Transfusion complications; Thalassemia diagnosis
Introduction
Thalassemia is a group of autosomal recessive hemoglobinopathies with high prevalence in populations originating from the Mediterranean, Middle East, and Southeast Asia [1,2]. Alpha and beta-thalassemia are molecularly heterogeneous resulting in a spectrum of phenotypic manifestations.
Two phenotypes determined by the degree of transfusion dependence define thalassemia [3]. Individuals with Transfusion Dependent Thalassemia (TDT) require life-long regular transfusions. Individuals with Non-Transfusion Dependent Thalassemia (NTDT) generally require infrequent or no transfusions, often diagnosed later in life. Although transfusion requirements may differ between TDT and NTDT, the risk for disease-related complications (hemosiderosis and alloimmunization) exist across both groups [4].
While individuals with TDT are cared for in either a hematologyoncology practice or recognized Thalassemia Treatment Center (TTC), characterization of care for NTDT individuals is less well defined [5]. Their unique needs may be unknown to the nonhematologists (primary care or emergency room physician) who participate in their care [6].
Methods
Using data from the Sickle Cell Data Collection Program [7] and Registry and Education for Hemovigilance in Hemoglobinopathy Transfusion Therapy (REdHHoTT), we attempt to better characterize the size of suspected NTDT and risk for transfusionassociated complications by focusing on those receiving intermittent transfusions in Georgia from 2007-2016. Our protocol for surveillance data collection was IRB exempt through Georgia State University; participant consent was not required.
Thalassemia case definition included individuals who had ≥3 healthcare encounters with a thalassemia diagnosis code (ICD9/10 codes) using hospital discharge data (inpatient and emergency department). Transfusions were identified by ICD9/10, CPT, revenue, or HCPCS code for red cell exchange or transfusion.
Results
Of 428 thalassemia patients identified, 187 (43.7%) had ≥1 transfusion. Sixty-nine (36.9%) of those patients received 1 transfusion; 118 had multiple transfusions. Of those multiply transfused, 57 patients (48.3%) received transfusions in multiple settings (30% of all transfused patients (Figure 1)). Of the 187 patients receiving ≥1 transfusion, women were transfused on average 50% more independent of the frequency of transfusion. Pediatric patients (0-19 years old) received the lowest number of transfusions (Figure 2).
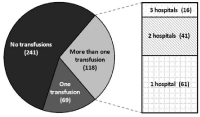
Figure 1: Transfusion Frequency and Location of Thalassemia Patients: 69
hospital sites provided at least one transfusion to 187 patients. Of the 118
patients receiving 1> transfusion, 57 received transfusions at multiple sites.
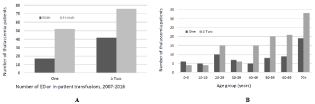
Figure 2: Transfused Thalassemia Patient Demographics, by Sex (a) and Age (b): 187 patients were identified who were transfused over the study period. Fiftynine
were male. 128 were female.
Discussion
We have identified a cohort of patients with likely NTDT who appear to be receiving transfusion care in inpatient and emergency settings in the state of Georgia. This suggests there may be individuals with thalassemia in Georgia receiving suboptimal therapy, as reflected in the location and frequency of their transfusion. These patients may not have a primary medical home. Efforts should be made to identify and educate provider groups included in this study on the current management guidelines for patients with thalassemia and the need for co-management with a hematologist. Previous studies show that a centralized regional or statewide transfusion database can enhance transfusion safety, particularly for patients seen at multiple institutions [8,9].
Conclusion
Many Georgia hospitals provide intermittent transfusions to low volumes of probable NTDT patients. These patients may receive transfusions at multiple sites that may not have access to their transfusion histories. Further study is needed to develop and validate methods for identifying NTDT patients who are not seen by TTCs but may need intermittent transfusion.
Limitations
The use of retrospectively collected billing data was based on ICD9/10 coding. The reliability of the provider to code a patient with a variation of beta thalassemia versus a patient with sickle cell beta-thalassemia may account for some of the patients in our cohort. We did not correlate underlying illness with transfusion episodes; therefore, some patients in our cohort may have required transfusion for anemia not attributable to thalassemia. This may account for the increased transfusion episodes among elder patient groups.
Disclosures
Registry and Education for Hemovigilance in Hemoglobinopathy Transfusion Therapy (REdHHoTT) is supported by the Centers for Disease Control and Prevention (NU58DD001138) and coordinated by the Georgia Health Policy Center. This content is the responsibility of the authors and does not necessarily represent the official views of the CDC or Department of Health and Human Services. The Sickle Cell Data Collection Program is supported by the CDC Foundation and CDC’s Division of Blood Disorders, with additional funding from the Doris Duke Charitable Foundation, Global Blood Therapeutics, Pfizer, and Sanofi.
References
- Cao A, Renzo G. Beta-thalassemia. Genetics in Medicine. 2010; 12: 61-76.
- Taher AT, Weatherall DJ, Cappellini MD. Thalassemia. Lancet. 2018; 391: 155-167.
- Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusiondependent thalassemias. Haematologica. 2013; 98: 833-844.
- Borgna-Pignatti C, Galanello R. Thalassemias and related disorders: quantitative disorders of hemoglobin. Wintrobe’s Clinical Hematology. Philadelphia: Lippincott Williams & Wilkins. 2004; 42: 1319-1365.
- Taher AT, Musallam KM, Cappellini MD, Weatherall DJ. Optimal management of β thalassaemia intermedia. British journal of haematology. 2011; 152: 512- 523.
- Taher AT, Radwan A, Viprakasit V. When to consider transfusion therapy for patients with non-transfusion-dependent thalassaemia. Vox Sanguinis. 2015; 108: 1-10.
- Sickle Cell Disease Collection Program, Registry, and Education for Hemovigilance in Hemoglobinopathy Transfusion Therapy (REdHHoTT) project sponsored by the Center for Disease Control and Prevention (CDC) unpublished data. Atlanta (United States). 2007.
- Delaney M, Dinwiddie S, Nester TN, Aubuchon JA. The immunohematologic and patient safety benefits of a centralized transfusion database. 2013; 53: 771-776.
- Harm SK, Yazer MH, Monis GF, Triulzi DJ, AuBuchon JP, Delaney M. A centralized recipient database enhances the serologic safety of RBC transfusions for patients with sickle cell disease. American Journal of Clinical Pathology. 2014; 141: 256-261.