
Research Article
Ann Hematol Oncol. 2021; 8(5): 1344.
Another Form of Modified, Highly-Active 6-Phosphofructo-1-Kinase in Cancer Cells
Kristl A, Camernik K, Avbelj Š and Legiša M*
National Institute of Chemistry, Slovenia
*Corresponding author: Matic Legiša, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
Received: March 08, 2021; Accepted: April 09, 2021; Published: April 16, 2021
Abstract
Enhanced glycolytic flux is a hallmarks of cancer cells. Posttranslational modification of the key regulatory enzyme of glycolysis, 6-Phosphofructo-1- Kinase (Pfk1) might trigger metabolic flux deregulation. In the cancer cells the human 85 kDa muscle type nPfk-M enzyme can be proteolytically cleaved to form highly-active 47 kDa shorter fragments that retain activity but become resistant to feed-back inhibition.
In several tumorigenic cell lines, no native 85 kDa liver type nPfk-L isoforms could be either found and only 70 kDa shorter fragments were detected by immune-blotting.
To learn more about the cancer-specific modified sfPfk-L enzyme, the truncated human sfPfk-L gene encoding 70 kDa fragments was inserted into the pfk null yeast S.cerevisiae cell. The recombinant modified enzyme showed higher affinity toward the substrate fructose-6-phosphate, reduced sensitivity toward the citrate and ATP inhibition in respect to the recombinant native PFK-L enzyme. Partially purified cancer-specific sfPfk-L fragments lacking the C-portion of the enzyme showed some instability under the diluted conditions in the buffer in respect to the tetrameric native nPfk-L enzyme. Growth characteristics of the yeast transformant encoding short sfPfk-L enzymes were similar to those encoding shorter sfPfk-M enzymes. No growth of the transformant with the sfPfk-L gene was observed on glucose but it grew faster than the transformant with the native human nPfk-L enzyme in a narrow ecological niche with low maltose concentration and 10 mM of ethanol in the medium.
Similar to modified 47 kDa sfPfk-M fragments, also the short 70 kDa nPfk- Lfragments might cause deregulation of the glycolytic flux in the yeast and in the cancer cells. In yeast, deregulated metabolic flux unbalances redox potential that results in reduced growth rate. However, the cancer cells beat the redox unbalance by rapid re-oxidation of redundant NADH that results in lactate formation while the growth rate remains high.
Keywords: Posttranslational modification; 6-Phosphofructo-1-kinase; Deregulated glycolytic flux; Tumorigenic cancer cells; Cancer; Lactate generation; Saccharomyces cerevisiae
Introduction
Almost a century ago, Otto Warburg noticed that rat liver carcinoma cells showed similar oxygen consumption in relation to normal cells, but neoplastic cells produced lactic acid even under aerobic conditions. This observation was in contrast to normal tissues that ceased to produce lactic acid in a fermentative way in the presence of oxygen (Pasteur effect). Nowadays, it is generally accepted that the cancer glycolytic phenotype is induced by oncogenic mutations that alter growth factor signaling [1]. Activated transcription complex HIF-1a is increased, which, in combination with transcription factor c-Myc, enhances the synthesis of the majority of glycolytic enzymes [2]. However, glycolytic flux in eukaryotic organisms is tightly controlled by allosteric enzymes; therefore, some modifications of the kinetics of regulatory enzymes must be involved in the metabolic changes during the transformation of normal cells into cancer cells.
The most complex control of glycolytic flux in mammalian cells is attributed to allosteric regulation of 6-Phosphofructo-1-Kinase (Pfk1), the last enzyme of the preparatory stage of glycolysis. Pfk1 catalyzes the phosphorylation of Fructose-6-Phosphate (F6P) to fructose-1, 6-bisphosfate, using MgATP as a phosphoryl donor [3]. Pfk1 is stimulated by fructose-2, 6-bisphosphate (F-2, 6-BP), ADP/ AMP, and ammonium ions, whereas citrate and ATP act as strong inhibitors [3]. During evolution, eukaryotic Pfk1 enzymes developed by duplication, tandem fusion, and the divergence of catalytic and effector binding sites of a prokaryotic ancestor [4]. However, the active site of the eukaryotic enzyme is located only at the N-terminal portion, while the allosteric ligand binding sites that developed during evolution by mutations and enable the fine-tuning of the regulatory enzyme are scattered at both the N and C termini.
We were the first to show that the human Pfk1 enzyme is subjected to posttranslational modification in cancer cells [5]. The C-terminal portion of the native 85-kDa enzyme is cleaved by a specific protease to form a highly active 47-kDa fragment. Newly formed, modified enzymes were resistant to the feedback inhibition by citrate and ATP, while F-2, 6-BP increased their activities to a level higher than that of the native enzymes. In several tumorigenic cell lines, only the shorter 47-kDa Pfk1 fragments and no native Pfk1 enzymes of approximately 85-kDa were detected using immunostaining against the muscle type nPfk-M isoenzyme [5].
In cancer cells, the enhanced synthesis of glycolytic enzymes by the transcription complex Hif-1a, in collaboration with transcription factor c-Myc, was proposed to cause enhanced glycolytic flux [2]. However, posttranslational modification of Pfk1 enzymes might be a more important phenomenon for the deregulation of glycolytic flux in tumors that, in combination with altered signaling mechanisms, essentially supports the fast proliferation of cancer cells.
In mammalian genomes, three different Pfk1 genes are present that enable the synthesis of proteins; they have the following molecular masses: muscle type (nPfk-M), 85,051 Da [6]; liver type (nPfk-L), 84,917 Da [7]; and platelet type (nPfk-P), 85, 596 Da [8]. In different human tissues, Pfk1 isoenzymes are differently expressed and different proportions of all three isoenzymes have also been found in different tumorigenic cell lines [9,10].
A different type of shorter fragment was observed in cancer cells. Several tumorigenic cell lines that originate from the metastatic phases of tumors were immune-stained with monoclonal antibodies raised against liver type nPfk-L. Only the 70-kDa fragments were observed, while 47-kDa fragments were absent. Again, the 85-kDa native protein could not be detected either. In the present paper, the presence of the shorter 70-kDa sfPfk-L fragments in some tumorigenic cell lines is described, and the changed kinetic characteristics in respect to the native enzyme are discussed.
Materials and Methods
Immunoblotting
The tumorigenic cell lines UOK 262, Colo 829, Jurkat, MDAMB- 231, Caco-2, UT-7 and Raji were either purchased from American Type Culture Collection (Manassas, Virginia, USA) or obtained through the courtesy of various investigators at National Institute of Biology (Ljubljana, Slovenia) or Josef Stefan Institute (Ljubljana, Slovenia). Mycoplasma testing has been performed of all cell lines before used in the experiments.
Before immunostaining, tumorigenic cell lines were incubated in the following media: Colo 829, Jurkat, and MDA-MB-231, and Raji cells were grown in RPMI 1640-Glutamax medium supplemented with 10% FBS, while UOK 262 and UT-7 were grown in DMEM with 10% or 20% FBS, respectively. Inoculation started with 1.105 cells/ mL and culture plates were incubated at 37°C and 5% CO2. The cells were harvested when the total number reached approximately 1.106 cells/mL. For the extraction, the cells were collected by centrifuging at 1500 rpm for 5 minutes and washed twice with cold PBS buffer. The cells were lysed by RIPA buffer that was left on ice for 20 minutes. Finally, cell debris was collected by centrifugation and supernatantcontaining solubilized proteins were preserved at -20°C until needed. Protein concentrations of the samples taken from different tumorigenic cell lines were determined by Coomassie Protein Assay kit (Pierce, Thermo Fisher; USA). For western blotting, samples of the cell line homogenates containing 20 μg of protein were separated on SDS-PAGE polyamide gel with 0.1 sodium dodecyl sulfate. The transfer of proteins to a nitrocellulose membrane was confirmed by Ponceau Red. The membrane was blocked with I-Block reagent (Tropix Inc.; Bedford, MA, USA), and washed and incubated with primary PFKL antibody (H-36) sc-292523 (Santa Cruz Biotech; Santa Cruz, CA, USA). Rabbit polyclonal IgG antibody was prepared after inoculation of the epitope corresponding to the mapping of amino acids 46-81 near the N-terminus of the nPfk-L of human origin. Subsequently, goat anti-rabbit-HPS (Abcam; Cambridge, UK) secondary antibody was used for detection. For the loading control, the lysates were analyzed using anti-β-actin antibody (C4)-HRP sc- 47778 (Santa Cruz, CA, USA).
Recombinant genes construction
The yeast strain HD56-5A (MATa ura3-52, leu2-3, 112his3-11, 15 MAL3 SUC2 GAL) was used as a wild-type strain, and its isogenic Pfk1, pfk2 null derivative, HD114-8D (MATa Pfk1::HIS3 pfk2::HIS3 ura3-52, leu2-3, 112his3-11, 15 MAL3 SUC2 GAL; [11], was used as a recipient of recombinant human genes.
The nPFKL gene encoding native human liver type nPfk-L and the sfPfk-L gene encoding the shorter 70-kDa sfPfk-L fragments were synthesized using the Gibson Assembly technique [12], after the codon usage of the human PFKL nucleotide sequence (CCDS33582.1) was adjusted for the expression in yeast cells. For the insertion of the genes, the p416-GPD vector [13] was first amplified by PCR using oligonucleotide 5’-TCA TGT AAT TAG TTA TGT CAC GCT TAC- 3’ as a forward primer and 5’-TCT TTA TCC GTC GAA ACT AAG TTC-3’ as a reverse primer.
Yeast transformants encoding native human nPfk-L and shorter 70-kDa fragments were designated for the nPFKL and sfPfk-L strains, respectively. Another transformant was constructed by inserting the vector carrying the sfPfk-L gene into the wild-type strain (HD56-5A). As a negative control, the HD114-8D strain was transformed using an empty p416 plasmid.
Growth of yeast transformants
Transformants were grown on Supplemented Minimal Medium (SMM), as reported previously [14]. The substrate contained synthetic dropout medium without uracil (Sigma-Aldrich; St. Louis, MO, USA) containing 2% glycerol and 2% ethanol as non-fermentable carbon sources. As a nitrogen source, yeast nitrogen base without amino acids or ammonium sulfate (Sigma-Aldrich; St. Louis, MO, USA) was used with the addition of glutamine (0.25 g/L).
Partial purification of recombinant PFKL proteins
Yeast transformants encoding nPfk-L and sfPfk-L enzymes as the only form of Pfk1 were grown in SMM-GE until an OD600 value of 0.6 was reached. The cells were harvested by centrifugation at 5000 x g for 5 min, and then washed with 50 mL of ice-cold 50 mM sodium phosphate buffer. The precipitate was frozen under liquid nitrogen and stored at -80°C until needed.
Frozen yeast cells were disrupted in a Mikro-Dismembrator (Sartorius AG; Gottingen, Germany). Cell-free homogenate was extracted with 10 mL of cold 50 mM sodium phosphate buffer (pH 7.8) containing 0.15 M glycerol, 1 mM DTE, 1mM PMSF, 1 mM EDTA, and 10 μL/mL of protease inhibitor cocktail (Sigma-Aldrich; Steinheim, Germany). The same buffer was used throughout the whole isolation procedure. Dissolved proteins in the supernatant formed after centrifugation at 16,000 x g for 20 min at 4°C and were precipitated with ammonium sulfate, and a fraction (between 45% and 75%) of the saturation was taken for further purification. After dissolving the precipitated proteins and desalting the sample on a SephadexTM G-25 column (GE Health Care; Piscataway, NJ, USA), the proteins were loaded onto an affinity column containing 1 mL of aminophenyl-ATP-Sepharose (Jena Bioscience; Jena, Germany) that had been previously equilibrated with extraction buffer. After the sample was applied to the column, unbound proteins were removed by extensive washing. Recombinant nPfk-L enzyme was eluted from the column with 1.5 mL of buffer containing 6 mM F6P and 1 mM ADP. Eluted enzyme was dialyzed overnight against a buffer containing 20% (v/v) glycerol and stored at 4°C. Due to the extreme instability of the sfPfk-L fragments, the affinity chromatography part of the purification process was omitted and enzyme kinetics were measured immediately after the desalting of the sample.
Enzyme assays
Pfk1 activity was measured spectrophotometrically at 340 nm (Lambda25 UV/VIS spectrophotometer, Perkin Elmer) as reported previously [15] using a coupled reaction system. Unless otherwise stated, in a final volume of 1 mL, the assay mixture contained 50 mM HEPES buffer (pH 7.8), 1 mM DTE, 100 mM KCl, 5 mM MgCl2, 0.2 mM NADH, 0.025 to 2 mM F6P, 10% v/v polyethylene glycol (PEG 6000), 0.9 U/mL aldolase (Sigma-Aldrich; Steinheim, Germany), 15 U/mL triosephosphate isomerase, and 15 U/mL glycerol-3-phosphate dehydrogenase (Sigma-Aldrich; Steinheim, Germany). Before use, the auxiliary enzymes were dialyzed overnight at 4°C against 50 mM HEPES buffer (pH 7.8) containing 1 mM DTE, with one change of buffer after 8 h. Different concentrations of citrate were added to the assay mixture just before the reaction was started by the addition of ATP to achieve a final concentration of 0.5 mM (if not specified differently). ATP concentration used enabled optimal Pfk1 activities, but was below the limit to induce inhibition. Concentration of the enzyme used in the kinetic assays was 0.2 μg/mL. All presented kinetic data are averages obtained from a minimum of three replicate measurements. Total protein concentrations of the samples were determined using a Bio-Rad protein assay (Bio-Rad; Hercules, CA, USA) with bovine γ-globulin as a standard.
Growth rate coefficients
To follow growth kinetics, the transformants were grown in 500 mL baffled Erlenmeyer flasks with 100 mL medium in a rotary shaker at 100 rpm and 30°C. The media were inoculated with a single-cell colony pre-grown on SMM with Glycerol-Ethanol (SMM-GE). For the different carbon source tests, identical SMM were used, but the nonfermentable sugars were replaced with glucose or maltose. Growth kinetics were monitored by measuring optical density (OD600) with a spectrophotometer (Lambda 25, Perkin-Elmer; Boston, MA, USA) and growth coefficients were determined as previously described [14].
Statistics analysis
Analyses were performed using GraphPad Prism version 6.00 for Windows (GraphPad Software, San Diego, CA) and the data were presented as the mean ± SD (n=3). Unpaired two tailed Student’s T-test was used for comparison between two groups. P<0.05 was considered to indicate a statistically significant difference.
Results
Detecting modified liver type sfPfk-L isoenzymes in metastatic tumor cell lines by immunoblotting
Previously, we found that the muscle type nPfk-M isoform can be predominantly cleaved into shorter 45 to 47-kDa fragments [5]. However, the presence of weak shorter 70-kDa fragments could be detected as well in some cell lines using polyclonal rabbit antibody raised against the specific epitope (CKDFREREGRLAA). A matching sequence of the nPfk-L isoform differs in four amino acid residues in respect to the nPfk-M epitope. To examine how liver type nPfk-L enzymes are modified in different neoplastic cell lines isolated from several metastatic tumors, polyclonal antibody raised against known epitope of the human nPfk-L enzyme were used for immunoblotting. The cell lines causing the following cancer types were tested: UOK 262, renal cell carcinoma; Colo 829, melanoma; Jurkat, acute T-cell leukemia; MDA-MB-231, mammary breast adenocarcinoma; Caco- 2, colorectal adenocarcinoma; UT-7 acute myeloid leukemia; and Raji, B-lymphocyte (Burkitt’s) lymphoma.
In the homogenate of all tested tumorigenic cell lines, the level of native 85-kDa nPfk-L was below the detection limit of the method. However, strong signals of approximately 70-kDa proteins were present in all tested cell lines. Faint 45-kDa fragments could be observed only in MDA-MB-231, UT-7, and Raji cells. Most importantly, no native nPfk-L enzymes with a molecular mass of 85- kDa could be detected in the extracts (Figure 1).
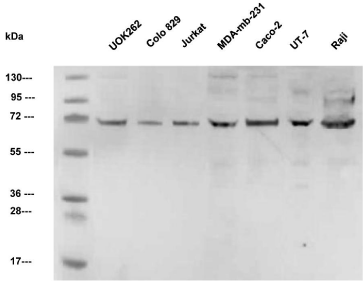
Figure 1: Western blots of homogenates of seven tumorigenic cell lines
immunostained by polyclonal nPfk-L antibodies. Only the shorter 70-kDa
sfPfk-L fragments can be observed, while no 85 kDa native nPfk-L enzyme
can be detected.
Preparing native liver type human nPFKL gene and truncated liver type human sfPfk-L gene
The nPFKL gene encoded the synthesis of the native nPfk-L protein of 780 amino acid residues with a molecular mass of 85,018,4 Da. A truncated sfPfk-L gene was designed to encode a shorter fragment of 648 amino acid residues with a molecular mass of 69, 942, 81 Da, lacking 132 amino acids at the C-terminus. The shorter sfPfk-L fragments were designed to end with the amino acid sequence SEKG, which was selected on the basis of the results published previously [15].
Expression of sfPfk-L gene encoding shorter 70-kDa fragments and nPFKL gene encoding 85-kDa active human enzyme in a pfk-null S. cerevisiae strain
All transformants, including the nPFKL and sfPfk-L transformants, were prepared in the pfk-null yeast strain (HD 114- 8D), and the wild-type (HD56-5A) with the inserted sfPfk-L gene, were first verified for the synthesis of active Pfk1 enzymes during the growth in non-fermentable glycerol/ethanol medium (SMM-GE). As anticipated, strong Pfk1 activity was detected in the cell-free extracts of the wild-type strain and both transformants with inserted nPFKL and sfPfk-L genes. A significant increase of specific Pfk1 activity was detected in the wild-type transformant with the inserted sfPFKM gene in respect to the genetically unmodified wild-type strain (Figure 2A). These data confirm that active native and shorter 70-kDa fragments can be synthesized in the yeast cells.
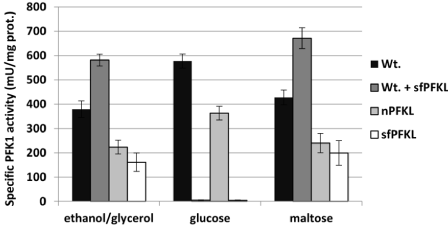
Figure 2: Specific Pfk-1 activities of the wild-type yeast and recombinant
enzymes. Specific Pfk-1 activities were measured in a cell-free extract of
the wild-type yeast, Saccharomyces cerevisiae, with the yeast nPFKL strain
encoding the human nPfk-L enzyme and the sfPfk-L strain encoding the
shorter 70-kDa sfPfk-L fragments as a sole form of Pfk1 enzyme in the cells.
In all tests, the strains were pre-grown in the non-fermentable sugars ethanol/
glycerol SMM-GE medium. For testing the effect of glucose and maltose,
the cells were transferred into the glucose or maltose SMM media for 15
minutes. Data are representative of three independent measurements and
are presented as means ± SD (n=3).
Later, specific Pfk1 activities were measured after the transfer of cells pre-grown in glycerol/ethanol medium into the SMM with 0.1% glucose or 0.1% maltose. After 15 minutes of incubation, the cells were collected by centrifugation and cell-free homogenates were prepared. Similar to the results observed previously with the sfPFKM strain [14], no Pfk1 activity was detected in the extract of the sfPfk-L strain after a short incubation in glucose. Also, no Pfk1 activity was observed in the extract of the wild-type transformant with the inserted sfPfk-L gene. However, all strains incubated in the 0.1% maltose medium showed Pfk1 activity (Figure 2B and 2C).
While the specific Pfk1 activities of the wild-type and nPFKL strain were lower during the growth in the maltose medium, as recorded during the incubation in the glucose, the combined wild-type/sfPfk-L strain showed higher activities than unmodified wild-type strain. The present data suggest that both wild-type yeast isoenzymes and the human shorter 70-kDa fragments were active. In the maltose SMM, specific Pfk1 activities in the extracts of both the nPFKL and sfPfk-L strains were about two-fold lower than those of the strains with the wild-type yeast enzymes (Figure 2C). These results suggest that the addition of glucose to the cells with shorter sfPfk-L fragments triggered a yet unknown safety mechanism that rapidly deactivated highly active sfPfk-L fragments.
Kinetic characteristics of the native nPfk-L enzyme and shorter 70-kDa sfPfk-L fragments
Recombinant human native 85-kDa nPfk-L protein and shorter 70-kDa PFKL fragments were partially purified from the nPFKL and sfPfk-L S. cerevisiae transformants after growth in the SMMGE with non-fermentable sugars. Both the native nPfk-L enzyme and the shorter fragments showed sigmoidal kinetics toward the substrate (F6P). However, marked effect of ATP concentration on kinetics of both enzyme forms was observed. While at low 0.2 mM ATP concentration similar affinities of both the native and shorter 70 kDa fragment were detected (Km 0.9±0.04 mM F6P), significantly higher affinity of the shorter fragment (Km 12.2±0.2) in respect to the native nPfk-L enzyme (Km 1.66±0.3) was noticed in the presence of 0,5 mM ATP in the measuring system. The Vmax values for both enzymes were reached at 2 mM F6P. However, at 1 mM ATP in the system, the inhibitory effect of the co-factor can be already observed with a reduced disparity of the affinities between the native (Km 1.97±0.13) and modified enzyme (Km 1.76±0.12). Higher saturation concentration (approximately 3.5 mM F6P) was also revealed for both enzymes at higher ATP concentrations (Figure 3).
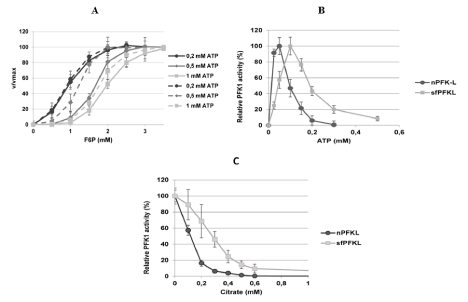
Figure 3: Kinetic characteristics of the purified human native nPfk-L and shorter 70-kDa sfPfk-L fragments. Shorter PFK-L fragments showed higher affinity toward
the substrate Fructo-6-Phosphate (FBP) in respect to the nPfk-L enzyme, however the highest difference was observed with 0, 5 mM ATP in the measuring system
(A). Both ATP (B) and citrate (C) acted as weaker inhibitors of the shorter 70-kDa sfPfk-L enzyme in respect to the native enzyme. Data are representative of three
independent measurements and are presented as means ± SD (n=3).
Besides, the native nPfk-L enzyme was more sensitive to ATP inhibition with an IC50 value of 0.1 mM ATP, while the shorter 70- kDa fragment’s activity was reduced by half only by 0.2 mM ATP concentration under the saturated F6P conditions. The shorter fragments were also less sensitive to citrate as an inhibitor, where twice as much citrate was needed to reduce the sfPfk-L activity by half (0.3 mM citrate) in respect to nPfk-L (0.15 mM citrate; Figure 3).
Similar to the human shorter 47-kDa sfPfk-M fragments, the shorter 70-kDa sfPfk-L fragment activities were unstable under the diluted conditions in a buffer (Figure 4). However, the calculated half turn over time of sfPfk-L fragments was 11.5 minutes, while the activity of sfPfk-M fragments was reduced by half within 5 minutes in the measuring buffer. The enzymes were more stable at cell-free extract that contained 10 mg proteins per mL and are expected to be fairly stable in the cell. To keep enzymes more stable during the kinetic measurements, 10% v/v polyethylene (PEG 6000) was added to the measuring buffer [5]. No instability of the human native nPfk-L enzyme could be detected under the identical buffer conditions.
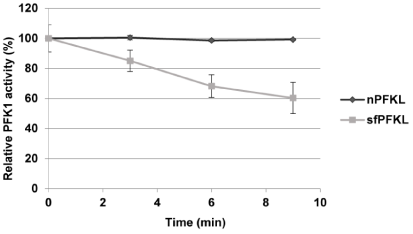
Figure 4: Enzyme stability: Specific enzyme activity of the partially purified
sfPfk-L fragment steadily decreased during the incubation in a buffer, while
no such effect was observed with the native nPfk-L enzyme. Data are
representative of three independent measurements and are presented as
means ± SD (n=3).
Growth of S. cerevisiae with inserted genes encoding native nPfk-L and shorter sfPfk-L fragments in supplemented minimal medium with maltose and ethanol
Both PFKL transformants were first tested for growth characteristics. The nPFKL strain grew well in the SMM with glucose or fructose, with a growth rate coefficient of about 0.25 (h-1), while no growth of the sfPfk-L strain was observed in such media. Similar to the sfPFKM yeast transformant encoding the muscle type human shorter 47-kDa fragments, the sfPfk-L strain grew on the maltose medium; however, 10 mM ethanol must have been added [14]. At the best growth conditions (0.1% maltose), the transformant-encoding liver type shorter 70-kDa isoenzyme reached a growth rate coefficient of 0.19 h-1 that surmounted that of the transformant encoding native nPfk-L (0.14 h-1) (Figure 5A) and the muscle type 47-kDa sfPfk-M isoenzyme (0.125 h-1) (Figure 5B). However, the modified liver type enzyme only enabled a sharp peak of maximal growth rate at 0.1% maltose concentration, but rapidly declined at lower and higher concentrations.
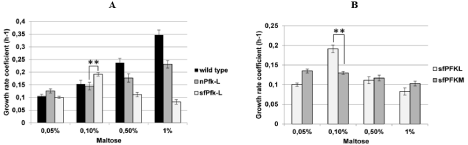
Figure 1: Growth rate coefficients of various yeast transformants in the maltose medium. While the sfPfk-L strain was unable to grow on a glucose medium, it grew even better than the wild-type nPFKL strain in a narrow niche in 0,1% maltose SMM medium (A). While the best growth coefficient of the sfPfk-L strain was observed at a peak in 0,1 maltose medium,, the sfPFKM strain encoding the 47 kDa muscle type sfPfk-M showed a steady increase of growth rate by decreasing concentrations of maltose in the medium (B). Data are representative of three independent measurements and are presented as means ± SD (n=3). The statistically significant differences (p<0.05, two-tailed t-test) in the increase of the growth coefficients of the sfPfk-L strain combined with the nPFKL strain at the peak in 0,1% maltose medium (A) and sfPfk-L strain combined with the sfPFKM strain at the peak in 0,1% maltose medium (B), are marked by (**).
In conclusion, shorter Pfk1 fragments with reduced regulatory capabilities of enzyme activities by the feedback inhibition enabled faster growth of the yeast cells at lower sugar (maltose) concentrations in respect to the highly controlled human native or yeast wild-type Pfk1 enzymes. The native enzymes with sophisticated regulation of enzyme activities were, therefore, capable of supporting the fast growth of the cells only at the higher sugar concentrations.
Although it is difficult to compare yeast and human energetic metabolisms and mechanisms that prevent detrimental ROS formation, these data suggest that under the specific physiological conditions, the cancer-specific liver type shorter 70-kDa sfPfk1 fragments might enable faster growth of the cells compared to the native Pfk1 enzymes and/or muscle type shorter 47-kDa fragments. Whether, intro-tumoral environmental conditions favor fast activity of the 70-kDa liver type sfPfk-L enzymes remains to be investigated.
Discussion
Recently, TAp73, a member of the p53 family, which is frequently overexpressed in cancer cells, was found to stimulate human liver type PFKL gene overexpression. Through this regulation, Tap73 was suggested to enhance glucose consumption and lactate generation, therefore, promoting the Warburg effect [16]. Interestingly, by testing a number of tumorigenic cell lines, only sfPfk-L enzymes with a molecular mass of approximately 70-kDa were found. This is consistent with our results of immunoblotting shown in Figure 1, where only the shorter 70-kDa sfPfk-L fragments were present, while the native 85-kDa enzymes were absent in the tested tumorigenic cell lines. However, there is only one gene (HGNC:8876) in the human genome encoding 780 amino acid residues long canonical nPfk-L enzyme of 85,018 Da (UniProtKB P17858-1). Tumorigenic cell lines used in the immunoblotting test shown in Figure 1 originate from the metastatic phase of aggressive tumors. Several authors reported that the Pfk-L enzyme was the predominant isoform in the later stage of tumor development in various cancer types [9,10].
There are strong indications that cancer-specific modified shorter sfPfk-L fragments might be formed by a posttranslational modification after the proteolytic cleavage of the C-terminus, similarly as detected earlier for the human muscle type nPfk-M enzyme [5,15]. Although 47-kDa fragments were spotted predominantly in several tumorigenic cell lines by immunoblotting using antibodies specific for the human muscle type nPfk-M enzyme, on some blots, a faint protein band corresponding to the shorter 70-kDa fragment was observed as well. However, no native 85 kDa nPfk-M enzymes were detected in any tested tumorigenic cell lines.
It has been suggested that human nPfk-M enzymes can be cleaved by serine protease in cancer cells. Specifically, human Kallikreinrelated protease 6 (KLK6), which is overexpressed in cancer cells, was able to cleave the native nPfk-M enzyme into a highly active shorter sfPfk-M fragment [15]. An increased amount of serine protease may cause auto-activation by cleavage of the pre-pro region in the cytosol where native Pfk1 enzymes are located. Overexpression of KLK genes has been reported to be stimulated by the constitutively active PI3K/ Akt/mTOR signaling pathway in breast and prostate cancer cells [17]. Proteins such as Cav-1 and Akt, which are known to be altered in colon cancer, also positively affect KLK6 expression [18].
In cancer cells, shorter 70-kDa sfPfk-L fragments might be formed after proteolytic cleavage by a serine protease as well. The target amino acid sequence SEGKGV, enabling cleavage after the lysine residue (K645) seemed to be the most appropriate target sequence for the formation of 69,942 Da sfPfk-L fragments. The SEGKGV sequence has also been predicted as a possible target for the Klk 6 [19]; however, the actual enzyme involved in the proteolytic modification of nPfk-L still needs to be determined.
Growth characteristics of the sfPfk-L strain in SMM with the addition of fermentative sugars, were similar to results to those of the sfPFKM strain encoding shorter 47-kDa sfPfk-M fragments [14]. Although both strains were unable to grow on glucose, they showed higher growth coefficients in respect to the wild-type strain in the SMM with a low maltose concentration and 10 mM ethanol. The uptake of maltose as a disaccharide is slower than the uptake of glucose [20], so the detrimental effect of highly active shorter fragment causing the deregulated glycolytic flux might be diminished by a maltose transporter. Besides, extracellular ethanol was shown to play an important role in rebalancing the NADH/NADPH ratio in yeast cells [14]. It could be suggested that both posttranslationally modified human Pfk1 isoforms, 70-kDa sfPfk-L fragments, and 47-kDa sfPfk-M fragments deregulate glycolytic flux and generate abundant NADH formation as a side effect. While no efficient mechanism for reoxidation of the surplus of NADH was obviously present in the yeast transformants, cancer cells solve this problem by the re-oxidation of NADH by the reduction of pyruvate to lactate [21].
Modified 70 kDa Pfk-L enzymes showed higher affinity toward the substrate and reached the Vmax activity earlier as the native nPfk-L enzyme. Unfortunately, the effect of fructose-2, 6-bisphosphate (F-2, 6-BP), a strong activator of Pfk1s, on the activity of both enzymes could not me measured because the substance is not commercially available any more. However, by previously conducted kinetic tests on human muscle type Pfk-M, a strong activation of the shorter 47 kDa fragments was observed by F-2, 6-BP. Although the activator increased the affinity of both enzymes toward the F6P as a substrate, the F-2, 6-BP also caused a marked increase in maximal velocity of the shorter fragments while no such effect could be recorded with the native nPfk-M enzyme [5].
In contrast to the shorter 47-kDa sfPfk-M fragments, the 70-kDa sfPfk-L fragments were not completely resistant to citrate inhibition. The IC50 value for citrate was determined to be 0.3 mM for the shorter 70-kDa sfPfk-L in respect to the native nPfk-L enzyme (IC50 0.15 mM). In humans, nPfk-L enzyme allosteric binding sites for citrate are scattered partly on the N-terminus (R35, R39, W70) and partly at the C-terminus (K556, V587, K616) [22]. While all citrate binding sites at the C-terminus are removed during the formation of the 47-kDa sfPfk-M fragments, all citrate binding sites remained at the 70-kDa sfPfk-L fragments. Somehow, reduced inhibition by ATP was recorded by measuring sfPfk-L kinetics as well. In mammalian Pfk1s ATP binding sites are represented by 6 amino acid residues that are scattered in the region between M201 to W462 [22]. However, two ATP binding sites (L766, E767) were later described to be located at the utmost C-terminus of the enzyme [23]; which are absent in the 70 kDa shorter sfPfk-L fragments.
Another difference between the modified sfPfk-M and sfPfk-L fragments was observed by measuring their stabilities under the diluted conditions in vitro. While the shorter sfPfk-M fragments form dimers [5], the sfPfk-L fragments most likely form tetrameric structures. From the model of the human Pfk1 crystal structure, amino acid residues responsible for binding two dimers into one tetramer were located at the Y634 and F638 positions of the nPfk-L enzyme [24]. Therefore, 70 kDa sfPfk-L fragments are less prone to the disintegration of monomers and, consequently, to the loss of enzymatic activity. As reported previously, the 252 amino acid residue prolongation at the C-terminus has been shown to stabilize the quarterly structure of all native eukaryotic Pfk1s [25].
To conclude, it is very likely that active shorter 70-kDa sfPfk-L fragments found in the several tumorigenic cell lines, significantly contribute to the deregulation of the glycolytic flux in the cancer cells. A similar phenomenon was previously described with shorter 47-kDa sfPfk-M fragments [5]. Highly active cancer-specific modified Pfk1 enzymes might, therefore, be an appropriate target for treatment with small-molecule inhibitors. By partial inhibition of modified cancerspecific Pfk1 isoforms, metabolic flux through glycolysis might be reduced to that of normal cells.
Acknowledgement
The authors wish to thank Dr. Toni Petan for kindly providing MDA-MB-231 cell culture and Dr. Nataša Kopitar Jerala for Raji cell culture.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism, Nat Rev Cancer. 2011; 11: 324-336.
- Huang LE. Carrot and stick: HIF-alpha engages c-Myc in hypoxic adaptation, Cell Death Differ. 2008; 15: 672-677.
- Dunaway GA. A review of animal phosphofructokinase isozymes with an emphasis on their physiological role. Mol Cell Biochem. 1983; 52: 75-91.
- Poorman RA, Randolph A, Kemp RG, Heinrikson RL. Evolution of phosphofructokinase--gene duplication and creation of new effector sites. Nature. 1984; 309: 467-469.
- Smerc A, Sodja E, Legisa M. Posttranslational modification of 6-phosphofructo- 1-kinase as an important feature of Cancer Metabolism. PLoS One. 2011; 6: e19645.
- Yamasaki T, Nakajima H, Kono N, Hotta K, Yamada K, Imai E, et al. Tarui, Structure of the entire human muscle phosphofructokinase-encoding gene: a two-promoter system., Gene. 1991; 104: 277-282.
- Elson A, Levanon D, Brandeis M, Dafni N, Bernstein Y, Danciger E, et al. The structure of the human liver-type phosphofructokinase gene. Genomics. 1990; 7: 47-56.
- Eto K, Sakura H, Yasuda K, Hayakawa T, Kawasaki E, Moriuchi R, et al. Cloning of a complete protein-coding sequence of human platelet-type phosphofructokinase isozyme from pancreatic islet. Biochem Biophys Res Commun. 1994; 198: 990-998.
- Vora S, Halper JP, Knowles DM. Alterations in the Activity and Isozymic Profile of Human Phosphofructokinase during Malignant Transformation in Vivo and in Vitro: Transformation- and Progression-linked Discriminants of Malignancy1. Cancer Research. 1995; 45: 2993-3001.
- Zancan P, Sola-Penna M, Furtado CM, Da Silva D. Differential expression of phosphofructokinase-1 isoforms correlates with the glycolytic efficiency of breast cancer cells. Mol Gen Metab. 2010; 100: 372-378.
- Raben N, Exelbert R, Spiegel R, Sherman JB, Nakajima H, Plotz P, et al. Functional expression of human mutant phosphofructokinase in yeast: genetic defects in French Canadian and Swiss patients with phosphofructokinase deficiency. Am J Hum Genet. 1995; 56: 131-141.
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases, Nat. Methods. 2009; 6: 343-345.
- Mumberg D, Müller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995; 156: 119-122.
- Andrejc D, Mozir A, Legisa M, Effect of the cancer specific shorter form of human 6-phosphofructo-1-kinase on the metabolism of the yeast Saccharomyces cerevisiae. BMC Biotechnol. 2017; 17: 41.
- Andrejc D, Legisa M. Kallikrein-related peptidase 6 can cleave humanmuscle- type 6-phosphofructo-1-kinase into highly active shorter fragments, Biochim. Biophys. Acta - Proteins Proteomics. 2018; 1866: 602-607.
- Li L, Li W, Chen T, Zou B, Zhao L, Wang H, et al. TAp73-induced phosphofructokinase-1 transcription promotes the Warburg effect and enhances cell proliferation. Nat Commun. 2008; 9: 4683.
- Paliouras M, Diamandis EP. An AKT activity threshold regulates androgendependent and androgen-independent PSA expression in prostate cancer cell lines. Biol Chem. 2008; 389: 773-780.
- Henkhaus RS, Roy UKB, Cavallo-Medved D, Sloane BF, Gerner EW, Ignatenko NA. Caveolin-1-mediated expression and secretion of kallikrein 6 in colon cancer cells. Neoplasia. 2008; 10: 140-148.
- Goettig P, Magdolen V, Brandstetter H. Natural and synthetic inhibitors of kallikrein-related peptidases (KLKs). Biochimie. 2010; 92: 1546-1567.
- Weusthuis RA, Pronk JT, van den Broek PJ, van Dijken JP. Chemostat cultivation as a tool for studies on sugar transport in yeasts. Microbiol Rev. 1994; 58: 616-630.
- Lunt SY, Vander Heiden MG. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu Rev Cell Dev Biol. 2011; 27: 441- 464.
- Kemp RG, Gunasekera D. Evolution of the allosteric ligand sites of mammalian phosphofructo-1-kinase., Biochemistry. 2002; 41: 9426-9430.
- Martínez-Costa OH, Hermida C, Sánchez-Martínez C, Santamaría B, Aragón JJ. Identification of C-terminal motifs responsible for transmission of inhibition by ATP of mammalian phosphofructokinase, and their contribution to other allosteric effects. Biochem J. 2004; 377: 77-84.
- Banaszak K, Mechin I, Obmolova G, Oldham M, Chang SH, Ruiz T, et al. The crystal structures of eukaryotic phosphofructokinases from baker’s yeast and rabbit skeletal muscle. J Mol Biol. 2011; 407: 284-297.
- Edelmann A, Kirchberger J, Heinisch JJ, Kopperschläger G. C-terminal modification of 6-phosphofructo-1-kinase from Saccharomyces cerevisiae and its influence on enzyme structure and activity, Biochem. Biophys. Res. Commun. 2002; 295: 992-999.