
Review Article
Ann Hematol Oncol. 2021; 8(8): 1361.
Clinical Review of Hereditary Angioedema: Diagnosis and Management
Weis M*
Department of Primary Care Medicine, Consolidated Troop Medical Clinic, Irwin Army Community Hospital, Fort Riley, KS
*Corresponding author: Mark Weis, Department of Primary Care Medicine, Consolidated Troop Medical Clinic, Irwin Army Community Hospital, Fort Riley, KS
Received: May 08, 2021; Accepted: June 09, 2021; Published: June 16, 2021
Abstract
Hereditary Angioedema (HAE) is caused by a deficiency in C1 esterase inhibitor and is characterized by sudden attacks of edema associated with discomfort and pain. The disease places patients at risk for disability and death if left untreated. Symptom severity and frequency can be extremely variable even among affected members of the same family. Attacks are not associated with inflammation or allergy, with most occurring secondary to trauma or stress. Swelling can affect any part of the body or multiple sites at once. Commonly affected areas include the extremities, genitalia, trunk, gastrointestinal tract, face, and larynx. Swelling typically worsens over 24 to 36 hours and resolves within 48 hours in less severe cases. Attacks result in 15 000 to 30 000 emergency department visits each year. Many of these emergency cases will undergo unnecessary surgeries or medical procedures due to misdiagnosis. The hallmarks of HAE recurrent episodes of swelling without urticaria, a family history of HAE, first attack in childhood, and worsening at puberty can be identified by a thorough family history, and the diagnosis can be confirmed by laboratory studies. Nevertheless, diagnosis may be delayed by 2 decades. We review available therapies and clinical characteristics that will both help clinicians diagnose HAE and distinguish among emergencies and nonemergency cases.
Keywords: Hereditary; Inherited; Angioedema; Swelling; C1 inhibitor; Bradykinin
Introduction
Hereditary Angioedema (HAE) is a debilitating disease with a significant risk for life-threatening episodes of swelling. Hereditary angioedema is characterized by sudden attacks of brawny, nonpitting edema that can cause both discomfort and pain [1]. The swelling typically affects the extremities, genitalia, trunk, gastrointestinal tract, face, and larynx. Death by asphyxiation has been reported in 30% of patients who had laryngeal attacks in the absence of treatment [2]. The possibility of asphyxiation from a first laryngeal attack is particularly disconcerting, especially to those patients who have family histories of HAE. Therefore, physicians as well as patients and their relatives need to develop and maintain a strong awareness of symptoms and appropriate interventions.
Hereditary angioedema is an inherited autosomal dominant disorder conferring a 50% chance of inheritance to offspring when one parent is affected (Figure 1), although 25% of cases arise from spontaneous mutations [3]. The disease is caused by a deficiency in C1 Esterase Inhibitor (C1 INH) [3]. Type 1 HAE is the result of a reduced quantity of circulating C1 INH and represents about 85% of cases [3,4]. Type 2 HAE is the result of dysfunctional circulating C1 INH and represents about 15% of cases [3,4]. A third type, HAE with normal C1 inhibitor activity, has recently been described [5]. This differs from classical HAE in that it is associated with normal C1 INH levels, and it is believed that attacks may be caused by defects in other pathways that result in bradykinin overproduction [6]. Type 3 HAE is seen primarily in women and, similar to types 1 and 2, appears to be estrogen dependent since affected women reported onset or worsening of symptoms with the use of estrogen-containing oral contraceptives [5]. However, estrogen dependency is not universal since cases of type 3 HAE have also been described in men [6].
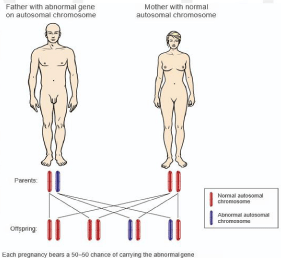
Figure 1: When one parent is affected, offspring have a 50% chance of inheriting HAE.
In autosomal dominant disorders, when one parent is affected, offspring have a 50% chance of inheriting the disorder.
Hereditary angioedema affects 1 in every 50 000 persons [7]. There are potentially 6000 HAE cases in the United States. Extreme phenotypic variability is observed among people with HAE even those from the same family. The frequency and severity of attacks are often unpredictable, and there may be little or no correlation between clinical symptoms and subtype of disease [4].
The mean age of HAE onset is approximately 11.2 years with about half of patients developing symptoms by age 10 years [8]. However, the diagnosis tends to lag considerably behind symptom onset due to the combination of low awareness of the disease in both the lay and medical populations and its symptom variability. The time between first HAE attack and accurate diagnosis ranges from 10 to 22 years [6]. In the absence of a diagnosis and appropriate treatment, patients may experience more frequent and more severe attacks and are at increased risk of death from laryngeal attacks.
Hereditary angioedema demonstrates no racial or sexual predilection. However, the clinical manifestations of HAE are more severe in women because fluctuating estrogen levels may worsen symptoms, causing women to seek medical care for symptoms more often than men [1]. While the available data do not show a conclusive link between estrogen levels and HAE symptoms, a larger percentage of affected women report worsening symptoms at puberty and with the use of estrogen-containing oral contraceptives [9]. In contrast, improved symptoms are reported by women taking progesterone only oral contraceptives [9].
Hereditary angioedema results in 15 000 to 30 000 emergency department visits annually [10], with 34% of patients who present with abdominal attacks undergoing unnecessary surgical or medical procedures due to misdiagnosis [11].
With proper diagnosis, patient education, and appropriate treatment, death and disability rates as well as health care costs may be substantially lowered. The following discussion of HAE will help clinicians distinguish among clinical presentations that constitute medical emergencies and those that are not emergencies. For patients presenting for the first time with symptoms suggesting HAE, it is critical that once the clinician confirms the diagnosis that he or she determine the subtype in order to offer disease specific therapy that both optimizes patient care and guides disease management.
Pathophysiology of HAE
Hereditary angioedema attacks are not associated with inflammation or allergy [12]. Trauma and stress are the most commonly reported triggers for HAE attacks, representing 54% and 43% of cases where the trigger can be identified, respectively [13]. However, attacks can occur without any obvious trigger. Hereditary angioedema attacks involve the activation of the contact, complement, and fibrinolytic cascades. C1 INH functions as a suicide substrate in these proteolytic pathways. Once C1 INH binds its target enzyme, the enzyme-substrate complex is cleared from the blood, and new C1 INH molecules must be produced to replace those that have been consumed. Figure 2 shows the sites of C1 INH binding in these proteolytic cascades. Since people with HAE have defective synthesis of C1 INH and have baseline plasma C1 INH levels between 5% to 30% of normal [2], the available C1 INH is quickly depleted once the cascades are activated via trauma, illness, or stress. People with this disease cannot generate C1 INH on pace with its depletion. Therefore, C1 INH deficiency and subsequent depletion allows the unregulated activation of proteolytic pathways and the unchecked production of bradykinin in the contact pathway [6]. The resulting excess bradykinin production increases vascular permeability, leading to localized edema, and is thought to be the primary cause of swelling in HAE [12].
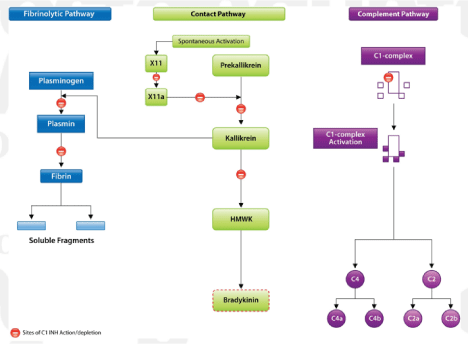
Figure 2: Plasma proteolytic pathways that deplete available C1 INH in an HAE attack.
The contact, complement, and fibrinolytic plasma proteolytic cascades are activated during HAE attacks. C1 INH is a suicide substrate in these pathways and is
consumed with each pathway iteration. Patients with HAE cannot produce adequate C1 INH to offset consumption in proteolytic pathways. Therefore, bradykinin
production in the contact pathway continues unchecked and causes swelling. This figure shows theoretical binding sites for C1 INH in these cascades. Adapted
with permission from N Engl J Med [6].
Clinical Presentation
Prodromal symptoms may precede an HAE attack and may include a tingling sensation in the soon-to-be-affected area [6]. Onethird of patients will have an erythema marginatum like eruption: a serpiginous, nonpruritic rash that is distinguished from urticaria [6]. Characteristic pink or red rings of an erythema marginatum like eruption commonly appear on the trunk and appendages prior to or during an attack. Once swelling begins, it typically worsens over 24 to 36 hours with slow resolution over the next 36 to 48 hours. Severe episodes can last for 3 to 5 days [14]. Swelling can occur in any part of the body and often affects multiple sites during an attack [3].
Nonemergency Cases
Extremity attacks are the most common, affecting 96% of patients [13]. These attacks often leave patients functionally disabled and may impede use of hands and feet for daily activities even walking. In addition, swollen joints can lead to discomfort and immobility. Extremity attacks rarely lead to hospitalization, but they do limit normal activity.
Urogenital attacks are common among patients with HAE and can result from sexual intercourse, childbirth, or other activities that place pressure on the genitourinary area, such as horseback riding [3]. These attacks usually lead to swollen genitalia and discomfort and, while not life threatening, may require treatment to relieve discomfort.
Emergency Cases
Abdominal attacks are the second most common among HAE attacks, affecting 93% of patients [13]. Abdominal attacks range in severity from mild to severe with intractable pain [2]. These attacks can be accompanied by vomiting, constipation, diarrhea, intestinal obstruction, and hypovolemic shock due to fluid loss to the abdomen [2]. The abdominal pain accompanying an attack is often spasmodic with increasing intensity [1].
Bowel sounds may vary from diminished to hyperactive [1]. The abdomen is usually protuberant with significant tenderness [1]. Occasionally, rebound tenderness and guarding may also be present [1].
Unfortunately, the combination of symptoms and physical findings during an abdominal HAE attack may mimic those of a surgical abdomen. Patients often present with intestinal obstruction complaining of severe cramps, nausea, and vomiting [3]. In the absence of an HAE diagnosis or clinical suspicion of HAE, misdiagnosis often results in unnecessary surgeries to remove normal appendices and gallbladders. While patients with abdominal attacks will benefit from pain relief, those with significant hypotension from fluid shift to the interstitium or peritoneal cavity require emergency care. These patients are best treated aggressively with fluid replacement to prevent hypovolemic shock, and often require narcotic analgesics until their symptoms abate. Imaging studies may be needed to differentiate between the surgical and nonsurgical abdomen in HAE patients presenting with abdominal pain. Therefore, it is important to treat each abdominal HAE attack associated with hypotension as a potential emergency.
Facial attacks are characterized by extreme swelling that causes temporary disfigurement [15]. These patients are at high risk of the attack extending to the larynx [15]. Half of patients with HAE will have at least 1 laryngeal attack in their life [2]. Laryngeal attacks are life-threatening and associated with significant mortality [2]. Patients with laryngeal involvement will require an emergency department visit and observation. They frequently require intubation or tracheotomy to prevent asphyxiation [13]. Therefore, it is imperative that all patients with facial HAE attacks be treated as emergency cases to prevent asphyxiation in the event of laryngeal extension.
Differential Diagnosis of HAE
Hereditary angioedema arises from many genetic mutations of any type that result in expression of dysfunctional C1 INH and the subsequent overproduction of bradykinin that causes swelling. Hereditary angioedema symptoms overlap with the symptoms of angioedema from other etiologies. Therefore, finding the root cause of the episodic edema is crucial to determining the appropriate management strategy.
The location of HAE attacks is highly variable, and the severity is unrelated to the magnitude of C1 INH dysfunction [4]. An untreated individual may experience >1 attack per week [6]. Alternatively, patients may have S 1 year of symptom-free intervals [8]. Precipitating events are often unpredictable. Only 39% of patients can identify the cause of their first
HAE attack [13]. However, known causes include physical trauma, surgical/medical/dental procedures, mechanical pressure during daily activities (typing, hammering), prolonged standing, infection, emotional stress, and some medications such as Angiotensin- Converting Enzyme (ACE) inhibitors, or estrogen-containing oral contraceptives [1,3,4,13].
Up to 25% of HAE cases arise from spontaneous genetic mutations [6]. In the absence of a family history to suggest HAE, initial and even recurrent presentations may be indistinguishable from acquired, idiopathic, or medication-induced angioedema. Hereditary angioedema can be distinguished from angioedema arising from other etiologies by a thorough patient history and laboratory evaluation. The clinical presentation for HAE will be characterized by nonpitted, nonpruritic subcutaneous or submucosal edema, possible nonpruritic serpentine erythematous rash, and possible migration of swelling to other sites [6]. A typical patient history may reveal the hallmarks of HAE which are recurrent swelling without urticaria, a family history of HAE, first attack early in life, and worsening symptoms at puberty [1,6]. Laboratory testing is indicated for patients with suspected HAE. Patients with HAE will typically have low C4 complement component protein levels with normal levels of C1q and C3 proteins [6]. Although C4 is generally a good screening test, there are instances in which patients’ C4 protein levels may be normal between HAE attacks, thus repeat testing during an acute attack may be helpful. Confirmatory testing of C1 INH antigenic and functional levels will allow definitive diagnosis and help distinguish between HAE subtypes [6]. Commercial laboratories offer C1 INH and C4 panels as separate tests, and some laboratories provide test selection and interpretation guidance tools, such as ARUP Consult® [16]. However, more specialized laboratories may offer an angioedemaspecific panel [17]. Table 1 summarizes the clinical symptoms and laboratory markers that help distinguish among HAE subtypes and other conditions with overlapping symptoms.
![]()
Angioedema Type
Clinical Description
Laboratory Studies [6]
Therapies
C4
Antigenic C1 INH
Functional C1 INH
C1q
C3
HAE type 1
Recurrent episodes of swelling in any part of the body without urticaria; family history of HAE; first symptoms in childhood; worsening symptoms at puberty; erythema marginatum-like
eruptions precede attacks in one
third of cases; swelling worsens
and resolves in 48 hours in less
severe cases [1,6].Low
Low
Low
Normal
Normal
C1 INH and danazol are FDA-approved therapies for routine prophylaxis [22,23]. Antifibrinolytics are used off label [27].
HAE type 2
Same as type 1.
Low
Normal
Low
Normal
Normal
HAE type 3
Same as type 1; possible estrogen sensitivity.
Normal
Normal
Normal
Normal
Normal
Acquired AE
Appears in middle age; family history of episodic swelling is absent [21].
Low
Low or normal
Low
Low
Low or normal
Treatment of possible lymphoproliferative disease [27]. Off-label antifibrinolytics or attenuated androdgens [27].
Idiopathic AE
S 3 episode of swelling in 1 year with no apparent cause; may be more prevalent in women; urticaria is present in half of cases [18].
Normal
Normal
Normal
Normal
Normal
Antihistamines, prednisone, or thyroid dysfunction therapy [27].
ACE inhibitor- induced AE
Closely resembles HAE; common sites include lips, tongue, face, but rarely bowels; typically presents in first three months of ACE inhibitor use; class effect adverse event [19].
Normal
Normal
Normal
Normal
Normal
ACE inhibitor discontinuation [20].
Allergic AE
Swelling typically with urticaria, possibly pruritic; possible anaphylaxis; caused by exposure to environmental allergen or drug; reactions have 24–48-hour duration [6].
Normal
Normal
Normal
Normal
Normal
Antihistamines, epinephrine, allergen avoidance [27].
Table 1: Clinical Characteristics of HAE, its Subtypes, and Other Angioedemas.
Idiopathic angioedema refers to S3 episodes of recurrent angioedema in a 1-year period with no apparent cause after comprehensive medical evaluation [18]. Idiopathic angioedema is the most common type of angioedema and is estimated to effect up to 20% of people worldwide at some point in life [18]. This angioedema may affect women more often than men [18]. The presence of urticaria in half of these patients helps distinguish idiopathic angioedema from that of other etiologies [18].
Angiotensin-converting enzyme inhibitor-induced angioedema is perhaps the second most common etiology due to the large number of patients worldwide who take these medications. The reported incidence of this condition varies from 0.1% to 6% [19]. African Americans appear to have an incidence of ACE inhibitor-induced angioedema 4 to 5 times that of white Americans [19]. The most common sites of ACE inhibitor-induced angioedema are the lips, tongue, and face, but in rare cases the bowel wall may be involved [19]. Angioedema associated with ACE inhibitor use most commonly presents in the first month of treatment [19]. However, it may not emerge for months or years and can remit and recur spontaneously if the ACE inhibitor is not discontinued [19]. This pattern of symptoms closely resembles HAE. For this reason, patients on ACE inhibitors presenting with angioedema should be evaluated to rule out HAE or acquired angioedema.
Angiotensin-converting enzyme degrades bradykinin. Therefore, the vascular effects of bradykinin are unregulated when ACE is inhibited [19]. Angiotensin-converting enzyme inhibitor-induced angioedema is a class effect adverse event. Those patients who experience this adverse event will likely continue to experience it regard- less of the ACE inhibitor used [19]. Discontinuation of ACE inhibitors is the best known therapy for this condition. Angiotensin Receptor Blockers (ARBs) appear to be a good alternative therapy in these patients, although a small percentage may continue to have symptoms while taking ARBs and ARBs will need to be discontinued [20]. Angiotensin-converting enzyme inhibitor use should be avoided in patients with HAE due to the decreased degradation of bradykinin upon ACE inhibitor use.
Acquired Angioedema (AAE) is typically the result of a lymphoproliferative disorder or autoimmune, neoplastic, or infectious diseases [2]. Its symptoms closely resemble those of HAE. Acquired angioedema can be differentiated from HAE by 2 clinical factors: 1) AAE appears in middle age, whereas HAE first manifests in childhood or adolescence; and 2) a family history of angioedema will be absent in the AAE patient [21].
Treatment
Patients with HAE will require individualized care to help reduce the impact of clinical symptoms on daily life. Prophylaxis is an important treatment option to help reduce the frequency and severity of attacks. For patients who have experienced significant disease burden due to the lack of a diagnosis or misdiagnosis, the transition to effective routine care will provide a welcome reduction in the frequency and severity of attacks.
Routine Prophylaxis
Only nanofiltered human C1 INH concentrate and the attenuated androgen danazol are approved for prophylaxis by the US Food and Drug Administration (FDA). The C1 INH CINRYZE™ (C1 esterase inhibitor [human]) is indicated for the routine prophylaxis against HAE attacks in adults and adolescents. CINRYZETM is administered by intravenous infusion of 1000 units every 3 to 4 days [22]. Upon administration of C1 INH, serum levels rise, thereby preventing the unregulated production of bradykinin via the contact pathway. In a randomized, double-blind, placebo-controlled crossover study [22] in 24 patients with a diagnosis of HAE and a history of S2 attacks per month, patients were randomized to either 12 weeks of C1 INH prophylaxis followed by 12 weeks of placebo or 12 weeks of placebo followed by 12 weeks of C1 INH prophylaxis. Twenty-two patients completed the study. While on treatment with C1 INH, patients experienced half the number of HAE attacks as those on placebo. Hereditary angioedema attacks among patients on active treatment were significantly less severe and significantly shorter in duration compared with HAE attacks occurring in patients on placebo. This study provided good evidence for clinically meaningful reductions in disease burden among patients treated with C1 INH. The most common adverse reactions were upper respiratory infection, sinusitis, rash, and headache [22].
Danazol is indicated for the prophylactic treatment of HAE in adults [23]. Danazol is supplied in 200-mg, 100-mg, and 50-mg capsules. The recommended initial dosage of 200 mg may be reduced by as much as 50% if patients experience durable prophylaxis [23]. Attenuated androgens are thought to increase endogenous C1 INH by hepatic stimulation [24]. The largest retrospective study of danazol efficacy involved clinical record analysis for 118 patients who received long-term danazol prophylaxis [25]. Danazol treatment was associated with an 83.8% reduction in the mean number of attacks per year. In all, 23.7% of patients reported being completely symptom free during danazol treatment, while the remainder continued to have attacks at a reduced frequency—14.4% had S 11 attacks per year. Adverse events were reported by 78.8% of patients. The most common adverse events were weight gain, menstrual irregularities, female virilization, headache, myalgia, depression, and acne. Less frequent but serious adverse events were also reported including 2 myocardial infarctions in the same 34-year-old man, stroke in a 58-year-old patient without known hypertension, 1 case of deep vein thrombosis, 1 case of acute pancreatitis, and 1 case of liver cell adenoma.
The antifibrinolytics, e-aminocaproic acid and tranexamic acid, have been used as prophylactic treatments in Europe. However, these agents carry significant risk for adverse events and are not approved for prophylaxis of HAE in the United States [6].
Procedural Prophylaxis
Dental and medical procedures are significant concerns for those with HAE. Angioedema can be precipitated by minor trauma to the tissue, such as dental work [4]. There are no FDA-approved therapies for procedural prophylaxis. However, consensus guidelines recommend off-label C1 INH administration 1 hour before a procedure at dosages of 500 units (patients weighing S 50 kg), 1000 units (patients weighing >50 kg and S 100 kg), and 1500 units (patients weighing >100 kg) [7]. According to the guidelines, the dose should be repeated at the time of the procedure and as necessary thereafter until the risk of swelling abates. Both danazol and Fresh Frozen Plasma (FFP) are recommended if C1 INH is unavailable. Typically 600 mg/day of danazol is administered for 1 week before and after the procedure.6 Two units of FFP 1 to 12 hours before the procedure is used as well [6], although the use of FFP increases the risk of bloodborne illnesses compared with other off-label therapies.
Acute Treatment
Berinert®, a plasma-derived C1 esterase inhibitor (human), was recently FDA approved for acute treatment of abdominal and facial HAE attacks in adults and adolescents [26]. In all, 124 patients were enrolled in a randomized, double-blind, placebo-controlled study. Patients were assigned to receive placebo or C1 inhibitor at dosages of 10 units/kg or 20 units/kg via intravenous infusion. Significantly shorter time to onset of symptom relief was reported by patients who received the 20 units/kg dose during an acute abdominal or facial HAE attack compared with patients receiving 10 units/kg C1 inhibitor or placebo. Berinert® is supplied in 500-unit vials with a recommended dosage of 20 units/kg. Commonly reported adverse events include increased pain associated with HAE, subsequent HAE attack, headache, abdominal pain, nausea, muscle spasms, pain, diarrhea, and vomiting. In the abscence of C1 inhibitor, off-label therapy consists of epinephrine or FFP. However, these agents have question- able efficacy [6]. Epinephrine may provide transient benefit, but its efficacy is not proven. Fresh frozen plasma carries the risk of worsening an attack by introducing additional contact pathway proteins. Other agents such as attenuated androgens, antifibrinolytics, corticosteroids, and antihistamines are inappropriate acute therapies because the attenuated androgens and antifibrinolytics are not quick acting, and corticosteroids and antihistamines are not effective for nonallergic reasons.
Investigational Agents
Agents under investigation for the treatment of HAE include a kallikrein inhibitor and a bradykinin receptor antagonist. The results of phase 3 studies and FDA review will determine their potential place as a therapeutic treatment option.
Summary
Hereditary angioedema is an inherited disease that dis- rupts patients’ lives and places them at risk for medical emergencies. Symptoms are caused by deficiency in C1 INH that allows local overproduction of bradykinin and subsequent swelling. Symptoms can vary drastically even among affected members of the same family. Low aware- ness of the disease and its symptom variability contribute to the lag in diagnosis. Some patients wait >2 decades for accurate diagnosis during which time they often experience periodic disability and an increased risk of death. Therefore, HAE should be suspected in patients who do not have a his- tory of significant allergies, who have a family history of recurrent swelling, and who present with recurrent episodes of swelling. In patients with suspected HAE, extremity and urogential attacks typically do not constitute emergencies, although they may cause significant discomfort or disability. However, laryngeal attacks, facial attacks, and abdominal attacks accompanied by hypotension warrant immediate referral to an emergency department. Food and Drug Administration-approved therapies exist for routine prophylaxis and acute treatment in adults and adolescents.
Acknowledgment
Editorial support was provided by ISC, LLC and was funded by ViroPharma, Inc.
Conflict of Interest Statement
Mark Weis, MD discloses a conflict of interest with Viro-Pharma, Inc.
References
- Frank MM. Hereditary angioedema: the clinical syndrome and its management in the United States. Immunol Allergy Clin North Am. 2006; 26: 653–668.
- Agostoni A, Aygoren-Pursun E, Binkley KE, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004; 114: S51–S131.
- Nzeako UC, Frigas E, Tremaine WJ. Hereditary angioedema: a broad review for clinicians. Arch Intern Med. 2001; 161: 2417–2429.
- Gompels MM, Lock RJ, Abinun M, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol. 2005; 139: 379–394.
- Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000; 356: 213–217.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008; 359: 1027–1036.
- Bowen T, Cicardi M, Bork K, et al. Hereditary angiodema: a current stateof- the-art review, VII: Canadian Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of Hereditary Angioedema. Ann Allergy Asthma Immunol. 2008; 100: S30–S40.
- Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006; 119: 267–274.
- Bouillet L, Longhurst H, Boccon-Gibod I, et al. Disease expression in women with hereditary angioedema. Am J Obstet Gynecol. 2008; 199: 484.e1-e4.
- Moore GP, Hurley WT, Pace SA. Hereditary angioedema. Ann Emerg Med. 1988; 17: 1082–1086.
- Agostoni A, Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore). 1992; 71: 206–215.
- Davis AE 3rd. Mechanism of angioedema in first complement component inhibitor deficiency. Immunol Allergy Clin North Am. 2006; 26: 633–651.
- Frank MM, Gelfand JA, Atkinson JP. Hereditary angioedema: the clinical syndrome and its management. Ann Intern Med. 1976; 84: 580–593.
- Zuraw BL. Hereditary angiodema: a current state-of-the-art review, IV: shortand long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008; 100: S13–S18.
- Bork K, Barnstedt SE. Laryngeal edema and death from asphyxiation after tooth extraction in four patients with hereditary angioedema. J Am Dent Assoc. 2003; 134: 1088–1094.
- ARUP Consult. The physician’s guide to laboratory test selection and interpretation: Hereditary angioedema. 2009.
- Frigas E, Park M. Quest Diagnostics. Angioedema panel, hereditary, comprehensive. Idiopathic recurrent angioedema. Immunol Allergy Clin North Am. 2006; 26: 739–751.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitorassociated angioedema. Immunol Allergy Clin North Am. 2006; 26: 725–737.
- Cicardi M, Zingale LC, Bergamaschini L, Agostoni A. Angioedema associated with angiotensin-converting enzyme inhibitor use: outcome after switching to a different treatment. Arch Intern Med. 2004; 164: 910–913.
- Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol. 2009; 155: 367–377.
- CINRYZE [package insert]. New York, NY: Lev Pharmaceuticals. 2008.
- Danocrine [package insert]. New York, NY: Sanofi-Synthelabo. 2003.
- Epstein TG, Bernstein JA. Current and emerging management options for hereditary angioedema in the US. Drugs. 2008; 68: 2561–2573.
- Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008; 100: 153–161.
- Berinert [package insert]. Kankakee, IL: CSL Behring. 2009.
- Hereditary Angioedema Association. Angioedema types. 2009.