
Review Article
Ann Hematol Oncol. 2022; 9(1): 1388.
Brain RAS in CNS Diseases: Beneficial Effects of Small Molecule Agonists and Inhibitors
Wright JW* and Harding JW
Department of Psychology, Department of Integrative Physiology and Neuroscience and Program in Biotechnology, Washington State University, Pullman, WA USA
*Corresponding author: John W Wright, Department of Psychology, Washington State University, P.O. Box 644820, Pullman, WA 99164-4820, USA
Received: January 11, 2022; Accepted: March 04, 2022; Published: March 11, 2022
Abstract
Neurodegenerative diseases are unrelenting, unforgiving and cruel given the long duration of patient suffering due to the impact of progressive damage within specific brain locations. In the case of dementias, there is a direct impact on memory and cognitive processing, and the loss of personal dignity and worth. Ultimately, the patient loses the ability to maintain basic hygiene placing attentional responsibilities on family members and support staff. With respect to neurodegenerative diseases of the eye, the patient must deal with progressive deleterious changes in vision resulting from retinal damage. This review discusses the role of the Renin-Angiotensin System (RAS) in cardiovascular disease, Alzheimer’s and Parkinson’s diseases, Type 2-induced dementia, depression, glaucoma, macular degeneration and diabetic retinopathy. We conclude with a consideration of the challenges posed regarding the development of new drugs designed to treat dementias, depression, and neurodegenerative diseases of the eye. The use of small molecule agonist and antagonist analogs of RAS components is discussed. These analogs can be configured to pass the blood-brain barrier and target relevant receptor proteins in specific brain structures or they can be applied topically to the eye to discourage increases in intraocular pressure, decreased retinal microvascular blood flow, tissue inflammation and oxidative stress as well as the accumulation of extracellular material (drusen) that can disrupt normal vision. Along with suggested drug development strategies, several important drug targets are identified in an effort to focus attention, and facilitate research efforts, to improve drug efficacy and thus provide better clinical outcomes for these patients.
Keywords: Renin-angiotensin system; Angiotensin receptors; Angiotensin receptor blockers; Dementias; Glaucoma; Macular degeneration
Abbreviations
Aβ: Amyloid-Beta Peptide; ACE: Angiotensin Converting Enzyme; ACEi: Angiotensin Converting Enzyme Inhibitor; AD: Alzheimer’s Disease; AH: Aqueous Humor; AMD: Age-Related Macular Degeneration; AP: Area Postrema; APA: Aminopeptidase A; APN: Aminopeptidase N; AngII: Angiotensin II; AngIII: Angiotensin III; AngIV: Angiotensin IV; ARB: Angiotensin Receptor Blocker; AT1: Angiotensin Receptor 1; AT2: Angiotensin Receptor 2; BBB: Blood-Brain Barrier; BDNF: Brain-Derived Neurotrophic Factor; Carb-P: Carboxypeptidase P; CBF: Cerebral Blood Flow; CVOs: Circumventricular Organs; DA: Dopaminergic ; DR: Diabetic Retinopathy; HGF: Hepatocyte Growth Factor; IGF-1: Insulin-Like Growth Factor; IOP: Intra-Ocular Pressure; IRAP: Insulin-Regulated Aminopeptidase; L-DOPA: Levodopa; LTP: Long-Term Potentiation; Mas: MAS1 Oncogene; MCI: Mild Cognitive Impairment; Met: N-Methyl-N’-Nitro-N-Nitrosoguanidine; MDD: Major Depressive Disorder; MPP+: MPTP Metabolite; MPTP: 1-Methyl-4-Phenyl- 1,2,3,6-Tetrahydropyridine; MRI: Magnetic Resonance Imaging; NGF: Nerve Growth Factor; NADPH: Nicotinamide Adenine Dinucleotide Phosphate; NO: Nitric Oxide; NTS: Nucleus of the Solitary Tract; PD: Parkinson’s Disease ; PAI-1: Plasminogen Activator Inhibitor 1; PO: Propyl Oligopeptidase; RAS: Renin-Angiotensin System; RPE: Retinal Pigment Epithelial Complex; SFO: Subfornical Organ; T2D: Type 2 Diabetes; VEGF: Vascular Endothelial Growth Factor
Introduction
The Renin-Angiotensin System (RAS) is recognized as one of the oldest hormone systems best known for its roles in regulating blood pressure and body water balance. In 1891 Robert Tiegerstedt and his student Per Bergan identified a pressor agent extracted from rabbit kidney tissues that they called “renin” [1]. Fifty years later, this finding led to the discovery of a vasoconstrictor agent isolated from ischemic kidneys of Goldblatt hypertensive dogs [2]. Page and Helmer [3] independently found the same molecule after injecting renin into intact animals. They also identified a “renin activator” later reported to be angiotensinogen [4]. This vasoconstrictor agent was eventually determined to be an octapeptide variously called, “renin substrate”, “hypertension” and “angiotensin”, ultimately termed Angiotensin II (AngII) [5-7].
This review initially describes the presently identified angiotensin ligands and their respective receptor subtypes. Angiotensin 1 and 2 (AT1 and AT2) subtypes have been well characterized and the AngII/ AT1 system is particularly important in the etiology of cardiovascular diseases [4,8,9]. The AT3 subtype was first isolated in mouse neuroblastoma cell cultures [10,11], but a separate gene has thus far not been sequenced in humans. The identity of the AT4 subtype has been controversial and will be discussed. Next, we will focus on the role of the brain Ang IV/AT4 receptor system in several neurodegenerative diseases. Additional diseases of the eye are identified as important targets requiring much additional research attention regarding the RAS and its relevance. Finally, recommendations are offered concerning drug development approaches in order to penetrate the blood-brain barrier and influence the brain RAS. Target diseases include dementias associated with Alzheimer’s and Parkinson’s diseases, Type II diabetes, as well as depression/neuroinflammation and diseases impacting the retina of the eye.
The Renin-Angiotensin System
The RAS is responsible for mediating several classic physiologies such as the regulation of systemic blood pressure and body water/ electrolyte balance, as well as a number of novel physiologies and behaviors including influences on sexual reproduction and behavior, Cerebral Blood Flow (CBF) and cerebroprotection, seizures, stress, depression, and memory [12,13]. AngII binds at the G-protein coupled AT1 receptor subtype [14-16]. Over the years the AngII/AT1 receptor system has been a major focus regarding the development of antihypertensive drugs and its role in promoting inflammation, oxidative stress and tissue remodeling [17,18]. These processes contribute to the “neuronal inflammation response” a key factor in the development of neurodegenerative diseases including Alzheimer’s Disease (AD) [19-21]. The biologically active angiotensin peptides are derived from the protein angiotensinogen (255 amino acids) via a cascade of enzymatic activity and include AngII (Asp- Arg-Val-Tyr-Ile-His-Pro-Phe), AngIII (Arg-Val-Tyr-Ile-His-Pro- Phe), Angiotensin IV (AngIV: Val-Tyr-Ile-His-Pro-Phe), Ang (1-7) (Asp-Arg-Val-Tyr-Ile-His-Pro) and Ang (3-7) (Val-Tyr-Ile-His-Pro) (Figure 1) [22-25]. Specifically, the decapeptide angiotensin I (AngI: Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu) is formed by renin (EC 3.4.23.15) acting upon the N-terminal of angiotensinogen. AngI serves as a substrate for angiotensin Converting Enzyme (EC 3.4.15.1) which is responsible for hydrolyzing the carboxy terminal dipeptide His-Leu to form AngII. AngII is converted to the heptapeptide AngIII by glutamyl aminopeptidase A (APA; EC 3.4.11.7) cleavage of the Asp residue at the N-terminal [26-28]. AngIII is acted upon by membrane alanyl aminopeptidase N (APN; EC 3.4.11.2) resulting in the cleavage of Arg to form the hexapeptide AngIV. AngIV can be further converted to Ang (3-7) by Carboxypeptidase P (Carb-P) and Propyl Oligopeptidase (PO) cleavage of the Pro-Phe bond. Angiotensin (1-7) is formed from AngII via Carb-P cleavage of Phe [29], by the monopeptidase ACE2 [30,31], and by ACE cleavage of the dipeptide Phe- His from Ang (1-9) [32].
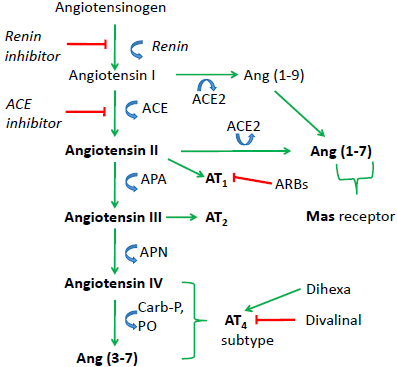
Figure 1: The angiotensinogen-renin-angiotensin pathway indicating
biologically active angiotensins (bold) enzymes, receptors and inhibitors
that mediate angiotensin physiologies and behaviors. Angiotensin II binds
predominantly at the AT1 receptor subtype and Angiotensin III at both the
AT1 and AT2 receptor subtypes. Angiotensin IV and Ang (3-7) bind at the AT4
receptor subtype. Angiotensin (1-7) binds at the Mas receptor.
ACE: Angiotensin Converting Enzyme; ACE2: Angiotensin Converting
Enzyme 2; AP-A: Aminopeptidase A; AP-N: Aminopeptidase N; ARBs:
Angiotensin Receptor Blockers; Carb-P: Carboxypeptidases; PO: Propyl
Oligopeptidase.
Ang II/AT1 and AngIII/AT2 receptor systems
The AT1 receptor subtype belongs to the superfamily of 7-transmembrane domain receptors and the AT1 gene is located in chromosome 3q and codes for a 359 amino acid protein (40-42 kDa) [14-16]. Signaling by the AT1 receptor is via phospholipase-C, -A2, -D-adenylate and calcium (L- and T-type voltage sensitive channels) [4,33,34]. The AT1 receptor is also coupled to intracellular signaling cascades involved in the regulation of gene transcription and protein expression that mediate cellular proliferation and growth in a number of target tissues, both peripheral and central. The AngII/ AT1 receptor system is a major player in cardiovascular functioning via direct inotropic influences on the heart and increases in vascular resistance [22,35]. Increased vascular resistance occurs due to direct vasoconstriction of vascular smooth muscle and indirect action via the brain resulting in sympathetic nervous system arousal, the inhibition of the baroreceptor reflex, and the release of the powerful vasoconstrictor arginine-vasopressin [36-38].
The AT2 receptor subtype is also a 7-transmembrane domain G-protein coupled receptor; however, it exhibits only about 32-34% amino acid sequence identity with the AT1 receptor [39,40]. This protein consists of a 363 amino acid sequence (40-41 kDa) [41] and is sensitive to AngII, but exhibits a higher affinity for AngIII [42]. This receptor is expressed in developing fetal tissues but decreases after birth and remains at low levels in adult tissues. The AT2 receptor subtype appears to modulate cell proliferation, cell differentiation, apoptosis, and regenerative processes and generally opposes actions initiated by the AngII/AT1 system [43,44]. It is important to note that the AT2 receptor can be upregulated during pathological conditions [45,46], although it is not clear to what extent this occurs in patients with neurodegenerative diseases.
Angiotensin IV/AT4 receptor system
Some time ago our laboratory, and others, discovered a binding site with nanomolar affinity for AngIV using bovine adrenal cortex membranes [47-49] and guinea pig hippocampal tissues [50]. The pharmacological profile of this receptor was shown to be distinct from the AT1 and AT2 subtypes. It was also determined that (125I)- AngIV binds at the AT4 site reversibly, saturably, and with high affinity. Binding was found to be insensitive to guanine nucleotides, indicating that this receptor protein is not G-protein-linked. Further, the AT4 receptor evidenced as a dimer, as seen in growth factors, with a molecular weight of 160-190 kDa as determined by reduced SDSpolyacrylamide gel electrophoresis [51]. This subtype is distributed within a number of brain structures with heavy concentrations in the hippocampus, nucleus basalis of Meynert, piriform cortex and neocortex, structures concerned with the mediation of cognition, learning and memory [52].
The AT4 receptor subtype has a positive influence on a number of physiological and behavioral functions including CBF, neuroprotection, synaptogenesis, Long-Term Potentiation (LTP), and memory consolidation and retrieval [53,54]. Jan Braszko and colleagues [55-57] were the first to report that intracerebroventricular injected AngIV facilitated exploratory behavior in rats tested in an open field, improved recall of passive avoidance conditioning and the memory acquisition of active avoidance conditioning. Members of our laboratory confirmed these memory results and further reported AngIV-induced dose dependent increases in CBF without significant changes in systemic blood pressure [58,59]. These effects could not be blocked by AT1 or AT2 receptor antagonists, but were prevented by pretreatment with the AT4 receptor antagonist divalinal-AngIV. Related to this, Naveri and colleagues [60] have shown that AngIV infusion restored CBF following subarachnoid hemorrhage. Further, Dalmay et al. [61] reported that AngIV infusion following pretreatment with the AT1 receptor blocker candesartan slightly decreased mortality at post-surgery Day 3 in the gerbil model of unilateral carotid artery ligation, but significantly decreased lisinopril-induced mortality. These results support the hypothesis that the activation of AT4 receptors contributes to cerebroprotection. This neuroprotective role for the AT4 receptor subtype is consistent with the notion that AngIV increases blood flow by a Nitric Oxide (NO)-dependent mechanism [59]. In agreement with this hypothesis Faure et al. [62] has shown that internal carotid artery administration of increasing doses of AngIV significantly decreased mortality and cerebral infarct size in rats 24 hours following embolic stroke due to the intracarotid injection of calibrated microspheres. Pretreatment with the AT4 receptor antagonist divalinal abolished this protective effect. Sequential cerebral arteriography indicated that AngIV caused the redistribution of blood flow to ischemic areas within a few minutes. It is hypothesized that AngIV may yield its neuroprotective effect against acute cerebral ischemia via an intracerebro-hemodynamic AT4 receptor-mediated NO-dependent mechanism. Most recently we have noted an interaction between AngIV-based analogs and the Hepatocyte Growth Factor (HGF)/Met system with evidence suggesting that the AngIV/AT4 receptor system coincides with the HGF/Met receptor system [54].
A potentially important advance in our understanding of the RAS was the finding that AngIV’s actions may be mediated in part by insulin-regulated aminopeptidase (IRAP: EC 3.4.11.3) and the hypothesis that this enzyme is the AT4 receptor [63,64]. IRAP is a Type 2 transmembrane protein of the gluzincin aminopeptidase family which includes homologous aminopeptidases such as aminopeptidases A and N [52,65]. IRAP co-distributes with the GLUT4 transporter [66,67]. The key substrates acted upon by this enzyme appear to be arginine-vasopressin and oxytocin [68]. It is proposed that the physiological and behavioral actions of AngIV are due to competitive inhibition of IRAP’s peptidase activity resulting in an extended half-life of AngIV and particularly oxytocin and vasopressin. IRAP has the capacity to cleave N-terminal amino acids from a number of peptides including met-enkephalin, dynorphin, oxytocin, arginine-vasopressin, lysine-bradykinin, neurokinin A1, somatostatin, neuromedin B, and cholecystokinin-8. IRAP has been variously identified as oxytocinase, cysteine aminopeptidase, placental leucine aminopeptidase, gp160, or vp165 [69]. Thus, IRAP inhibition by Ang IV results in the potentiation of several pro-cognitive endogenous peptides including arginine-vasopressin, oxytocin, somastatin and cholecystokinin-8 [70]. However, Albiston and colleagues [68] reported that IRAP gene knock-out mice revealed impaired performance on memory tasks rather than enhanced performance as predicted. This finding casts some doubt concerning the relative importance of IRAP’s role in the potentiation of memory formation and retrieval. In a subsequent report, these investigators measured an absence of IRAP in members of a postnatal forebrain neuron-specific IRAP knockout mouse line. As predicted these animals’ revealed dysfunctions in spatial and object recognition memory at three months of age. The results suggested that the presence of IRAP in the postnatal brain may be necessary for normal memory functioning [71].
Members of our laboratory have questioned the notion that IRAP is the AT4 receptor [42] and offered another possibility, namely the Hepatocyte Growth Factor (HGF)/Met receptor system [12,52,72-76]. This came about based on a search for a molecular target with a chemical structure similar to AngIV, and behavioral and physiological functions in agreement with those discovered for the AngIV/AT4 receptor system. A partial match was seen with the protein angiostatin, and a related member of the plasminogen family HGF. Functions associated with the HGF/Met system overlap with those mediated by the AngIV/AT4 system including facilitated memory consolidation, augmented neurite outgrowth, hippocampal LTP and calcium signaling, dendritic arborization, facilitation of CBF and cerebroprotection, seizure protection, and facilitated wound healing [52,53,74]. This prompted the hypothesis that AngIV and AngIV analogs may function via the HGF/Met system. We have reported that the AT4 receptor antagonist Norleual-AngIV inhibited HGF binding to Met and HGF-dependent signaling, proliferation, invasion, and scattering [72]. Norleual-AngIV’s mechanism of action regarding this ability to act as a Met receptor antagonist is by inhibiting the dimerization of HGF which serves as a prerequisite to Met receptor activation [72,77]. HGF dimerization is a necessary step in order to bind to and activate the Met receptor [78,79]. This dimerization process is dependent upon a short HGF domain located between its N-terminal and first Kringle domain referred to as the “hinge region” (Figure 2) [13,79]. Members of our laboratory have shown that a hexapeptide, designed to mimic the hinge region, bound to HGF with high affinity and blocked HGF dimerization [77]. We hypothesized that AngIV and AngIV analogs bind at this hinge region and facilitate HGF activation thus leading to increased Met receptor activation. There is now evidence that this appears to be the case [80].
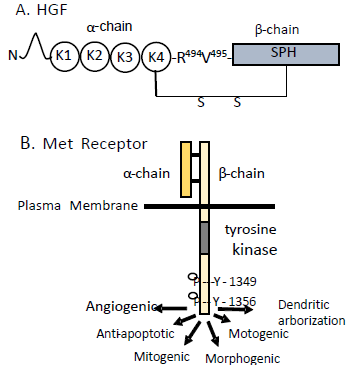
Figure 2: A) Structure of HGF consisting of a a-chain (69 kDa) accompanied
by four Kringle domains and a β-chain (43 kDa) including a Serine Proteinase
Homology (SPH) domain, linked by disulfide bonds (S). High affinity binding
sites are located at the N-terminal domain and the first Kringle domain of the
a-chain. B) Structure and basic functions of the Met receptor consisting of
a a-chain (50 kDa) and a β-chain (140 kDa) linked by disulfide bonds. HGF
binds to the Met receptor resulting in tyrosine phosphorylation leading to the
activation of a number of biological activities including those listed plus antineuroinflammation
and inhibition of oxidative stress, increased cerebral blood
flow and synaptogenesis, and facilitated long-term potentiation and memory.
Clearly it is not necessary that this issue by resolved in favor of one hypothesis or the other, since it has been shown that AngIV and AngIV analogs interact with both IRAP [65] and the HGF/Met system [13]. It is likely that these systems work together such that competitive inhibition of IRAP functions to extend the half-life of AngIV allowing it a longer duration to bind at the HGF/Met receptor system. In addition, as mentioned earlier AngIV can be converted to Ang (3-7) which also binds at the AT4 receptor. This friendly controversy concerning the identity of the AT4 receptor, plus new important findings about the Ang (1-7)/Mas receptor system, has served to reinvigorate research interest in the brain RAS. Members of our respective research groups have cooperated on past projects [81] and hopefully this collaboration can continue in the future.
Angiotensin (1-7)/mas receptor system
Ang (1-7) binds at the Mas receptor that is also G-protein-coupled and has been shown to counteract peripheral organ inflammation and fibrosis, increase glucose utilization, and decrease insulin resistance [82-84]. The Mas receptor is present in brain structures associated with memory and cognition including hippocampal and piriform cortices [85]. Consistent with these observations Ang (1-7) has been shown to facilitate LTP (a presumed building block of memory formation) in the CA1 region of the hippocampus via activation of the Mas receptor [82]. The reader is referred to the following articles and reviews concerned with detailed characterization of the angiotensin receptor subtypes, and the Mas receptor [4,31,52,83,86,87].
Independent brain RAS
During the 1970s Detlef Ganten and colleagues reported the presence of renin and AngII in the dog brain resulting in the recognition of an intrinsic independent brain RAS [88-90]. This amazing discovery, along with subsequent research findings, revealed that the brain RAS is one of many local RASs that mediate intracellular communication among various cell types (a paracrine role) as well as same cell types (an autocrine role) [91,92]. These local systems, for example the heart, liver, intestine, pancreas, ovary, uterus, testis, and eye cooperate in the regulation of cell differentiation, growth, proliferation, metabolism, apoptosis, tissue inflammation, fibrosis, hemodynamics and hormone secretion [93-95]. Following Ganten’s discovery other investigators reported that intracerebroventricular injections of AngII in animal models produced potent increases in blood pressure via activation of AT1 receptors located in Circumventricular Organs (CVOs), particularly the Subfornical Organ (SFO) and Area Postrema (AP), that project to the paraventricular and supraoptic nuclei of the hypothalamus [37,96]. Microinjections of AngII into the SFO and organum vasculosum of the lamina terminalis also elicited reliable elevations in blood pressure [97,98]. The pressor response due to circulating AngII was shown to be mediated primarily by the SFO and AP. The absence of a Blood-Brain Barrier (BBB) at these CVO sites permits penetration by this peptide and other circulating hormones. AngII also activates cardiovascular centers in the medulla. Target structures include the Nucleus of the Solitary Tract (NTS), AP and anterior ventrolateral medulla [38]. In particular, the AP detects blood-borne AngII as does the NTS which influences the baroreceptor reflex [99,100]. AngII delivered to the anterior ventrolateral medulla increases blood pressure by facilitating the sympathetic nervous system and catecholamine release from the adrenal medulla [100,101-103]. In summary, an overactive RAS can result in a hypertensive state accompanied by reduced cerebral blood flow, elevated oxidative stress and a pro-inflammatory response, resulting in cognitive dysfunction [13].
Cardiovascular Disease
Nearly fifty years ago it was reported that minor structural modifications of AngII yielded peptides capable of acting as antagonists at the AT1 receptor subtype. Two of these compounds, saralasin (Sar1, Ala8-AngII) and sarile (Sar1, Ile8-AngII) were evaluated in clinical trials but were dismissed primarily because of their peptidic structures [104-107]. Even so these peptides have been useful as research tools that highlighted the importance of the RAS, and particularly the AT1 receptor, in mediating systemic blood pressure [65]. Such studies led to the development of the first non-peptidic Angiotensin Receptor Blocker (ARB) losartan, in 1995 [108]. Since then, several additional ARBs have been introduced and successfully taken through clinical trials including candesartan eprosaran, olmesartan, telmisartan and valsartan [109,110]. Azilsartan is the most recent to receive FDA approval in 2011 [111]. All are antihypertensive drugs designed to block the AT1 receptor subtype and reduce blood pressure. In addition, both losartan and candesartan have been shown to facilitate cognitive processing in elderly hypertensive patients, an important observation [112-115].
The zinc-binding thiol compound captopril was the first Angiotensin-Converting Enzyme Inhibitor (ACEi) to be developed as an anti-hypertensive drug [116]. The major side effects of taste disturbances and skin rash were eliminated in most patients by the introduction of enalopril [117]. Several ACEi followed including benazepril, lisinopril, perindopril, quinapril, ramipril [65,118]. These drugs are designed to inhibit the conversion of AngI to AngII and reduce activation of the AT1 receptor subtype resulting in a sustained decrease in systemic blood pressure. It has been shown that captopril and perindopril influence not only the peripheral but also the central RAS [119]. Along these lines, mild to moderate male hypertensive patients treated with captopril indicated improved mental acuity, less sexual dysfunction and an improved sense of wellbeing [120]. Amenta and colleagues [121] reviewed clinical trials results concerning the influence of anti-hypertensive treatments on cognitive processing in hypertensive patients. They concluded that ACE inhibitors improved cognitive functioning independent of blood pressure effects and superior to β-blockers and diuretics. Further, ACE inhibitors have been reported to facilitate cognitive performance and reduce the occurrence of vascular dementia following hemorrhagic or ischemic cerebrovascular accidents.
Stabilization of cognitive performance by ACE inhibitors has also been noted in patients with Mild Cognitive Impairment (MCI) [122,123]. Hajjar et al. and others [124,125] have reported a slowed rate of cognitive decline in Alzheimer’s Disease (AD) patients placed on ACE inhibitors. In contrast, Sudilovsky et al. [126] reported ceranopril to have no effect on cognitive functioning in AD patients; while Khachaturian et al. [127] found ACE inhibitors to be the only anti-hypertensive drug to indicate a slightly increased incidence of AD. For thoughtful reviews concerning the development of these drugs and their chemical structures and targets beyond cardiovascular disease the reader is referred to the following papers [65,83,128].
Diseases that Impact Memory and Cognition
Early on a role for AngII in the facilitation of memory and cognition was proposed [42,51,87,129-131]. However, subsequent animal studies indicated that intracerebroventricular delivery of AngII interfered with performance on most memory tasks used with animal models [12]. This finding agreed with reports that ARBs improved cognitive processing as mentioned earlier. But if AngII acting at the AT1 receptor interfered with memory, and blockade of this receptor improved memory, what was the mechanism responsible for this memory facilitation? A majority of recent results point to AngIV interacting with the AT4 receptor subtype as the source of memory improvement [13]. These collective results can be explained as follows. Blockade of the AT1 receptor subtype prevents memory interference and permits unbound endogenous AngII to be converted to AngIV, which then binds at the AT4 receptor. This notion is supported by the observation that ACE inhibitors enhance cognitive processing in both humans [123,132] and animal models [133]. The resulting increases in AngI levels due to inhibition of ACE are likely converted to Ang (1-9) to Ang (1-7) and then to Ang (3-7). Ang (3-7) has been reported to act as an agonist at the AT4 receptor subtype [134]. AngIV analogs such as Nle1-AngIV, have shown promise in overcoming the memory impairments evidenced by several animal models of AD. Intracerebroventricular (icv) treatment with Nle1-AngIV reversed memory deficits due to: 1) application of the cholinergic muscarinic receptor antagonist scopolamine [135]; 2) kainic acid-induced lesions of the hippocampus [136]; 3) perforant path knife-cuts [136]; 4) embolic stroke due to carotid artery injection of microspheres [62]; and 5) ischemia resulting from transient fourvessel occlusion [137,138]. This latter finding is important given the strong possibility that cerebral hypoperfusion acts as a precursor to the development of MCI followed by AD [139]. Consistent with these behavioral results [125I] AngIV has been autoradiographically localized within structures known to mediate cognitive processing including neocortex, hippocampus, and the basal nucleus of Meynert [50,87,140].
Alzheimer’s Disease
Patient numbers
Approximately 5.5 million people in the U.S. are diagnosed with Alzheimer’s Disease (AD) [141,142] and more than 16 million worldwide [143]. In 2017 it is estimated that 6.08 million Americans were afflicted with AD. This number is predicted to reach 15 million by 2060 [144], and three times that worldwide [145]. Treatment and care costs for the U.S. patients is estimated at $70-100 billion [146,147] and worldwide in excess of $600 billion [148]. Without a breakthrough in treatment, these numbers of AD patients and associated costs may overwhelm our health care systems. There is also growing concern over concussion-induced cortical damage seen in children and adults who participate in contact sports such as American football (chronic traumatic encephalopathy), boxing, martial arts, and soccer, as well as our service men and women who have experienced combat associated concussions [149.150]. Evidence indicates that repeated concussions may encourage MCI [151].
FDA approved drugs
Despite intensive research efforts, only two categories of drugs have been approved by the FDA to treat AD, and only one in the past 20 years. Cholinesterase inhibitors such as Razadyne, Exelon, Cognex and Aricept disrupt the degradation of acetylcholine thus extending the half-life and availability of this neurotransmitter acting at central cholinergic muscarinic and nicotinic receptors [53,152- 154]. A second approach utilizes an N-Methyl-D-Aspartate (NMDA) receptor antagonist, Namenda (memantine HCl), to limit glutamate excitotoxicity and resulting neuronal damage [152,155,156]. These drugs have demonstrated limited ability to delay the symptoms of AD and none prevent disease progression. However, both Roche and Eli Lilly have experienced monoclonal antibody Aβ drug failures in advanced clinical trials resulting in trial terminations [157,158]. Tau aggregation inhibitors are also being tested designed to discourage the formation of neurofibrillary tangles [159].
It should be noted that Biogen’s controversial “plaque buster” monoclonal antibody drug to amyloid-β (Aβ), aducanumab (BIIB- 037), was approved by the FDA in June 2021. However, the Scientific Review Committee voted to deny approval citing the presence of brain swelling, and some brain bleeds, in several clinical trials participants. This decision is currently under FDA review.
Biomarkers of alzheimer’s disease
AD patients present extensive distributions of senile plaques and neurofibrillary tangles accompanied by neuroinflammation, oxidative stress-induced damage and a pronounced loss of synaptic connections predisposing neuronal apoptosis [160]. Plaques composition includes aggregates of amyloid-beta peptide (Aβ) due to a significant elevation in the production of neurotoxic Aβ (1-42) [161,162]. The Aβ (1-42) peptide oligomerizes resulting in neuronal toxicity. Neurofibrillary tangles are characterized by aggregated hyperphosphorylated tau protein. These proteins normally act to stabilize microtubules but in AD patients, they contribute to a loss of neuronal structural integrity ultimately impacting synaptic connections.
The goal of providing an effective treatment for AD has been elusive in part due to the multifactorial characteristics of the disease process and difficulty in identifying reliable biomarkers [163-165]. Presently established diagnostic indicators of AD are present in other forms of dementia including frontotemporal, vascular, diffuse Lewy body, corticobasal, dementia due to Parkinson’s disease and HIV infection, as well as normal aging [166-170]. It has been speculated that the pathology associated with AD may initiate many years prior to the occurrence of clinical symptoms [139,154,171]. Thus, considerable effort is being directed toward the development of early detection techniques via monitoring saliva, serum, cerebrospinal fluid, neuroimaging biomarkers, and behavioral measures of cognitive dysfunction [172-177]. Reliable detection at the earliest signs of AD related pathology could permit treatment many months or even years ahead of symptoms. Research must continue with the ultimate goal of preventing neuronal damage and preserving memory and cognitive functioning. However, efforts must also focus on the interim strategy to delay neuron losses in memory associated brain structures including the hippocampus, nucleus basalis of Meynert, piriform and neocortices. A drug designed to slow pathology, and thus major symptoms, would extend the patient’s quality of life and significantly reduce health care costs. De la Torre [178] has calculated that delaying disease onset by 5 years would reduce the number of diagnosed patients by upwards of 50%.
Parkinson’s Disease
Symptoms and treatments
James Parkinson first described this disease in 1867 and Parkinson’s Disease (PD) now affects approximately 1.5% of the world’s population over 65 years of age [179]. PD is characterized as a progressive loss of brain Dopaminergic (DA) neurons in the substantia nigra pars compacta. The striatum is the primary projection field of substantia nigra DA neurons. The loss of DA synthesis and release results in insufficient stimulation of dopaminergic D1 and D2 receptors throughout the striatum [180-182]. Decreased availability of DA triggers a triad of symptoms including bradykinesia, tremorsat- rest, and rigidity. Discussion continues over the pathogenesis of PD with arguments in favor of both genetic and environmental factors. There is growing evidence from animal models and PD patients that neuroinflammatory processes, likely triggered by reactive oxygen species, damage mitochondrial membrane permeability, enzymes and mitochondrial genome, leading to DA cell death [183,184].
Levodopa (L-DOPA) has been shown to be effective at controlling motor symptoms in the majority of patients but is ineffective regarding non-motor symptoms. Current treatment strategies to relieve motor symptoms include DA replacement via L-DOPA (the precursor of DA), DA receptor agonists, monoamine oxidase B inhibitors, and catechol-O-methyltransferase inhibitors (to protect the DA that is formed). As the disease progresses periods of decreased mobility, dyskinesia, and spontaneous involuntary movements complicate treatment (Marsden, 1982). These motor dysfunctions are currently treated with the DA receptor agonists apomorphine and levodopa, and surgical techniques including pallidectomy and deep brain electrical stimulation [185-187]. Progressive DA neurodegeneration may also impact additional non-dopaminergic neurotransmitter systems including noradrenergic, cholinergic, and serotonergic [188]. As a result, non-motor symptoms may develop including depression, sleep disturbances, dementia, and autonomic nervous system failure [189-191]. L-DOPA continues to be the most efficacious oral delivery treatment for the control of motor symptoms [192]. Unfortunately, L-DOPA is reasonably ineffective at combating non-motor symptoms [189]. Thus, current research efforts are directed at controlling these additional symptoms, as well as the development of new strategies designed to offer neuroprotection and overall disease reversal benefits. Attaining the goals of slowing and hopefully reversing the rate of DA neuron loss may also result in the protection of non-DA neurotransmitter systems.
The RAS and parkinson’s disease
A relationship between the brain renin-angiotensin system and Parkinson’s disease was first suggested by Allen and colleagues [193]. These researchers measured decreased angiotensin receptor binding in the substantia nigra and striatum in post mortem brains of PD patients. This can be explained by the fact that in addition to the systemic RAS described earlier there are local RASs present in many tissues including the brain [194]. These local systems also synthesize angiotensins that mediate the action of many substances including cytokines and growth factors involved in cellular growth, apoptosis, and inflammation [195,196]. Locally formed AngII binding at AT1 receptors activates Nicotinamide Adenine Dinucleotide Phosphate (NADPH)-dependent oxidases that are a source of superoxide (O2) which is upregulated in diabetes, hypertension and atherosclerosis [197-201]. Activation of the AT1 receptor also results in the synthesis of chemokines, cytokines, and adhesion molecules, all important in the migration of inflammatory cells into regions of tissue injury [202]. Autoradiographic studies have identified AT1 receptors in substantia nigra DA neuron cell bodies and terminal fields in the striatum in a number of mammalian species including humans [16,203,204], with humans evidencing the highest levels [193].
Several studies support an important role for ACE in PD. ACE is present in the nigra-striatal pathway and basal ganglia structures [205-207]. PD patients treated with the ACEi perindopril showed improved motor responses to the DA precursor 3,4-dihydroxy-Lphenylalanine [208]. Relative to treatment with perindopril, elevated striatal DA levels were measured in mice [209]. ACE inhibitors have been shown to inhibit bradykinin metabolism and thus modulate inflammation and induce blood vessel dilation [210], which are key factors in neurodegeneration. Activation of the AT1 receptor subtype by AngII activates NADPH-dependent oxidases, a significant source of reactive oxygen species [45,211]. Treatment with ACE inhibitors has been shown to offer protection against the loss of DA neurons in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) animal models [209,212], as well as the 6-OHDA rat model [213]. The likely mechanism underlying this ACEi-induced protection is a reduction in the synthesis of AngII acting at the AT1 receptor subtype [214,215].
In light of the above reports, it follows that if AngII activation of the AT1 receptor subtype results in facilitating the NADPH oxidase complex and the formation of free radicals, then blockade of the AT1 receptor should serve a protective function. This appears to be the case given that treatment with AT1 receptor antagonists have been shown to protect DA neurons in both the 6-OHDA [216] and the MPTP-models [217]. AT1 receptor antagonists have been shown to reduce the formation of NADPH oxidase-derived reactive oxygen species following administration of 6-OHDA. A recent study designed to evaluate the relationship between treatment with antihypertensive drugs and the risk of developing a first-time diagnosis of PD, found no association with ACE inhibitors or AT1 receptor antagonists, while treatment with calcium channel blockers was associated with a reduced risk of Parkinson’s disease diagnosis [218]. It should be noted that there may be methodological concerns with this investigation [219].
A potential role for AngIV and the AT4 receptor in PD has been investigated [220]. A genetic in vitro PD model was used consisting of the a-synuclein over-expression of the human neuroglioma H4 cell line. Results indicated a significant reduction in a-synuclein-induced toxicity with losartan treatment combined with the AT2 receptor antagonist PD123319, in the presence of AngII. Under these same conditions, AngIV was only moderately effective. However, these authors did not use a metabolically stable AngIV analog nor did they employ an AT4 receptor antagonist in combination with AngII or AngIV.
In summary, experimental work suggests that treatment with an AT1 receptor antagonist may offer some protection against the risk of developing Parkinson’s disease [214]. However, results from an observational study concerning antihypertensive treatment and the risk of Parkinson’s disease were disappointing regarding treatment with ARBs and ACE inhibitors [218]. Much additional work must be conducted to better understand the relationship among brain angiotensin receptors and ligands, neuroinflammation and reactive oxygen species, as related to Parkinson’s disease.
Relationship Among RAS, Hepatocyte Growth Factor and Parkinson’s Disease
Neurodegeneration and aging
Aging represents a major risk factor in predisposing individuals to neurodegenerative diseases [221-223]. The neurodegeneration accompanying aging is dependent in part upon oxidative stress, neuroinflammation, and microglial NADPH oxidase activity. Each is of significant importance regarding DA neuron loss [224,225]. Activation of AT1 receptors by AngII has been shown to facilitate DA neuron degeneration by activating microglial NADPH oxidase [225]. The activation of AT1 receptors by AngII failed to cause DA neuron degeneration when microglial cells were absent [211]. Of related importance, Zawada and colleagues [226] recently reported that nigral dopaminergic neurons respond to neurotoxicity-induced superoxide in two waves. First, a spike in mitochondrial hydrogen peroxide was measured three hours following treatment with an MPTP metabolite (MPP+). Second, by twenty-four hours following treatment hydrogen peroxide levels were further elevated. Treatment with losartan suppressed this nigral superoxide production suggesting a potentially important role for ARBs in the treatment of PD. Further, AngII binding at the AT1 receptor increased DA neuron degeneration initiated by subthreshold doses of DA neurotoxins by stimulating intra-neuronal levels of Reactive Oxygen Species (ROS) and neuroinflammation by activation of microglial NADPH oxidase [199,201,227,228].
AT1 receptor subtype blockade
From the above observations it follows that AT1 receptor blockade should have a neuroprotective effect on DA neurons in PD patients as demonstrated in animal models [217,220]. Less obvious is the likelihood that AT1 receptor blockade results in accumulating levels of AngII which is converted to AngIII and then to AngIV. This conversion cascade has been shown to occur intracellularly [229]. In fact, this conversion of AngII appears to be necessary for DA release to occur in the striatum [230]. Thus, an intriguing alternative explanation concerning these AT1 receptor antagonist results is that the increased endogenous levels of AngIV facilitate activation of the HGF/Met receptor system and in turn neuroprotection of DA neurons. In this way AngIV may act, in combination with AT1 receptor blockade, to protect DA neurons.
In agreement with the above hypothesis, HGF has been shown to positively impact ischemic-induced injuries such as cardiac [231] and hind limb ischemia [232,233]. HGF has also been shown to eliminate hippocampal neuronal cell loss in transient global cerebral ischemic gerbils [234], and transient focal ischemic rats [235]. Date and colleagues [236] have reported HGF-induced improvement in escape latencies by microsphere embolism-cerebral ischemic rats using a circular water maze task. These authors measured reduced damage to cerebral endothelial cells in ischemic animals treated with HGF. Shimamura et al. [237] have shown that over-expression of HGF following permanent middle cerebral artery occlusion resulted in significant recovery of performance in the Morris water maze and passive avoidance conditioning tasks. Treatment with HGF was also found to increase the number of arteries in the neocortex some 50 days following the onset of ischemia.
In sum, these results suggest a role for the HGF/Met receptor system in cerebroprotection and are consistent with the notion that AngIV increases blood flow by a NO-dependent mechanism [238]. There have been reports of increasing doses of AngIV via the internal carotid artery significantly decreasing mortality and cerebral infarct size in rats twenty-four hours following embolic stroke due to the intracarotid injection of calibrated microspheres [137]. Pretreatment with the AT4 receptor antagonist Divalinal, or Nω-Nitro-LArginine Methyl Ester (L-NAME), abolished this protective effect. Sequential cerebral autoradiography indicated that AngIV caused the redistribution of blood flow to ischemic areas within a few minutes. Thus, AngIV may yield its cerebral protective effect against acute cerebral ischemia via an intracerebro-hemodynamic Met receptormediated NO-dependent mechanism.
Type 2 Diabetes
Case numbers
At present, there are approximately 29 million diabetic patients in the U.S. with 1.4 million new cases diagnosed each year. Of these 90% are Type 2 leaving the remainder as Type 1. World-wide the number of Type 2 diabetic patients is estimated to be 380 million (International Diabetes Federation) [239]. This number is anticipated to increase to 430+ million by 2050. Over time a significant number of these patients, perhaps as high as 10%, will develop AD-like symptoms [240-242]. Both Type 2 Diabetes (T2D)-induced dementia and AD are now considered “metabolic diseases” in that they evidence impairments in insulin responsiveness and glucose utilization. In the case of AD this impaired response to insulin encourages, in part, brain inflammation, oxidative stress, the accumulation of β-amyloid protein within neurons, tau hyperphosphorylation, and the loss of synaptic connections resulting in neuronal apoptosis in memoryrelated structures [243-246]. Hallmarks of T2D include brain insulin resistance and impaired insulin signaling that can initiate abnormal glucose metabolism, inflammation and oxidative stress responses, mitochondrial dysfunction, and vascular damage [241,244]. Thus, T2D and AD patient’s exhibit common biomarkers and the resulting T2D-induced cognitive impairments create long-term consequences with similar impacts on the patient, family members, care givers, and health care providers as AD [240,247]. Since T2D and AD patients share many biomarkers, and the presence of T2D accelerates the possible onset of AD-like symptoms [240,241], it is reasonable to look for predisposing factors common to both diseases. An overlooked contributor to the metabolic dysfunction seen in both AD and T2D is the role of the RAS. An argument can be made that T2D is facilitated by the onset of organ vulnerability to diabetic-induced hyperglycemic injury and over activity of local RASs [248-250].
AngII levels and oxidative stress
It has been known for some time that hyperglycemia induces oxidative stress; however, elevated AngII tissue levels have also been shown to act as an oxidative stress-inducer [251,252]. Thus, elevated AngII concentrations in diabetic tissues may exacerbate hyperglycemia-induced oxidative stress damage [248,249]. As a result, oxidative stress appears to both underlie, and be the result of, patho-biochemical mechanisms of diabetic-induced tissue damage [250]. For example, the retina and kidney have been reported to have over-active local RASs during episodes of hyperglycemia [253-255]. Elevated pro-renin levels have been measured in the vitreous of the eye in diabetic patients with proliferative retinopathy [256]. Some older patients with this disorder evidenced increased vitreous AngII levels [257]. Further, there is evidence that vitreous AngII levels are positively correlated with the degree of retinopathy [256]. There is a strong correlation between organs vulnerable to diabetic-induced hyperglycemic injury (e.g. kidney and retina) and the over activation of local RASs [258,259]. Increased AngII concentrations in these tissues appear to promote end-organ damage in at least two ways: 1) by activating AT1 receptor proteins thus inducing changes in local blood flow and tissue hydration; and 2) exacerbating hyperglycemicinduced oxidative stress, elevated polyol and hexosamine pathway variability, and facilitating glycation end-products. Thus, the use of drugs to inhibit the RAS has become an important treatment approach to control diabetic nephropathy, and to a lesser extent retinopathy.
In support of this hypothesis is the finding that the inhibition of the RAS with ACE inhibitors, or ARBs, in diabetic nephropathy rats reduced oxidative stress [16]. Several clinical trials have evaluated the efficacy of RAS blockade with diabetic patients. One noteworthy trial focused on young Type 1 diabetic patients evidencing vascular superoxide overproduction (an early sign of angiopathy) due to hyperglycemia-related dysfunctional intracellular antioxidant enzyme production [260]. This dysfunction was reversed by treatment with the ARB irbesartan. Further, the ARBs candesartan and R-147176 (a sartan with low affinity for the AT1 receptor subtype) appear to exert direct antioxidant influences presumably independent of AT1 receptor blockade [261]. Thus, these drugs show promise with regard to protecting against diabetic-induced end-organ damage. However, they do not protect against T2D-induced dementia.
Type 2 diabetes and dementia
The diagnosis of T2D presents a major risk factor in the development of dementia. Type 2 diabetes is generally associated with aging and occurs at the rate of 20% in people over 65 years of age [246,262]. As previously indicated T2D is characterized by a number of metabolic disorders including cellular insulin resistance, compromised glucose utilization, and chronic inflammation. These dysfunctions facilitate cellular damage to kidneys, eyes, vasculature as in coronary artery disease, neuropathy and other end-organ damage [244]. The recognized metabolic syndrome associated with T2D include hyperinsulinemia, hypercholesterolemia, and hyperglycemia may encourage brain neuron losses resulting in structural atrophy. In addition, these neuronal pathologies are shared with AD patients [244]. It has been estimated that T2D patients may suffer a two-fold increase in the life time risk of dementia [263]. At least 10% of the current world-wide population of T2D patients evidence dementia characteristics. An intensive evaluation of 100,000+ cases of dementia revealed that the presence of diabetes resulted in a 60% increased risk of dementia in both men and women [264]. The relative risk of vascular dementia for T2D diagnosed women was 2.34-time controls, and 1.49 for men. The risk of nonvascular dementia was elevated 1.53 for women and 1.49 for men. These analyses argue that world-wide there are an additional 30+ million T2D dementia patients to be added to the 47 million AD patients [265].
It has now become accepted that the treatment of T2D patients with ACE inhibitors or ARBs reduces activation of the RAS with resulting reductions in hypertension and oxidative stress, and also impacts local HGF/Met receptor systems. Along these lines treatment with an ACE inhibitor reduces the formation of AngII; however, the resulting increase in the nonapeptide, D-Asp1,AngI, leads to the cleavage of aspartate by APA, followed by conversion to AngIII with the cleavage of histidine and leucine via carboxypeptidases activity, and then to AngIV via APN cleavage of arginine [21]. This resulting elevation in circulating AngIV levels activates dimerization of HGF followed by increased binding at the Met receptor thus optimizing hepatic and cellular insulin responsiveness. A similar outcome would be anticipated with ARB treatment of T2D patients. Thus, the positive response of T2D patients treated with an ARB [19,20,65] may be due to an excess of AngII that cannot bind at the AT1 receptor subtype. This excess AngII is converted to AngIII, and then to AngIV and Ang (3-7). Both AngIV and Ang (3-7) are capable of facilitating dimerization of HGF, which then activates Met receptors in the pancreas and elsewhere. Activation of Met receptors in turn increment insulin production and facilitate cellular insulin responsiveness, with accompanying reductions in hyperglycemia-induced oxidative stress and end-organ damage. Unfortunately, these elevations in AngIV and Ang (3-7) do not prevent T2D-induced dementia. Our best guess is that both AngIV and Ang (3-7) have difficulty penetration the BBB and thus are not significantly impacting the brain. In addition, members of our laboratory determined years ago that the half-life of AngIV is in the range of 10-20 seconds. We did not know about the importance of Ang (3-7) at that time so did not test it.
In summary, a relationship exists between the development of Type 2 diabetes and the likelihood of neurodegeneration resulting in Alzheimer’s disease-like symptoms. A complete understanding of the factors underlying this neuropathology has not been forthcoming. However, it appears that components of the RAS, specifically the AngII/AT1 receptor system, are activated by T2D, and in turn contribute to processes characteristic of AD including neuroinflammation, oxidative stress, reduced cerebral blood flow, destructive tissue remodeling and damage to the cellular mechanisms underlying memory consolidation and retrieval. These same pathologies have been identified in patients afflicted with T2Dinduced dementia [240], and a role for the RAS has been suggested [246]. An AngIV analog may be an effective treatment for T2Dinduced dementia [54]. This suggestion is bolstered by the finding that the HGF/Met receptor system has been identified as important in diabetes 266,267].
Depression and Neuroinflammation
Major depressive disorder
Major Depressive Disorder (MDD) is a common form of mental disorder affecting approximately 15% of the U.S. population at least once during lifetime [268]. Approximately 17 million American adults experience one or more episodes of depression in a year [269]. In addition, episodes of depression are experienced by about 2% of children and 5% of adolescents [270]. The likelihood of depression increases with age particularly among those with functional disabilities, and/or physical and cognitive illness [271-273]. About 10% of community/residence-seniors report symptoms of major depression [272,273]. The pathophysiology of adult depression is complex with contributing factors that include CNS and peripheral systemic factors, while Alzheimer’s disease, Parkinson’s disease, stroke, alcohol/drug addiction, and other chronic diseases, are recognized risk factors [274-276]. In particular, cancer, cardiovascular disease, metabolic and endocrine dysfunction are often associated with depression [277,278].
RAS and depression
The first suggestion that the brain RAS is important in depression came with the observation that captopril induced an anti-depressant effect in hypertensive patients that also suffered from depression [279- 282]. There had been previous hints concerning this relationship from animal studies. Specifically, rats treated with antidepressants revealed decreased water intake induced by peripherally or centrally injected isoprenaline, either in the presence or absence of a a2-adrenoceptor antagonist [283,284]. Further testing indicated that each of the antidepressant drugs fluoxetine, desipramine, and tranylcypromine, reduced AngII-induced dipsogenicity in rats [285,286].
Captopril treatment has also been shown to protect animals against the forced swim induction method of learned helplessnessinduced depression. This protocol requires the animal to swim within a small pool of water that has no escape. Eventually the animal stops swimming and becomes immobile. The next day it assumes swimming immobility significantly sooner than during the initial trial. In each subsequent test day, the latency to evidence immobility decreased, i.e. “learned helplessness”. Pretreatment with captopril reduced immobility by mice equivalent to treatment with the antidepressants imipramine or mianserine [287]. Learned helplessness induced by foot shock in rats could be prevented by pretreatment with captopril to the same effect as imipramine [288]. Under both protocols the protective effects of captopril were reversed by naloxone, suggesting that the ACE inhibitor was exerting its antidepressant effects, at least in part, via opioid receptors. In addition, this effect is also dependent upon the brain RAS since pretreatment with losartan provided protection from immobility in the forced swim test [289,290]. These results suggest that antidepressants exert their positive effects to some degree by inhibiting the brain RAS. The precise mechanism(s) of this inhibition remains to be determined. There is recent evidence that the chronic infusion of AngII may facilitate depression in adult C57BL/6 mice [291]. These animals were prepared with subcutaneous osmotic pumps and infused over a 21-day period. The mice evidenced depression-like behaviors when tested using forced swimming and tail suppression tasks. This depressive state could be reversed with imipramine or telmisartan. The authors hypothesized that AngII acts via microglia activation of the hippocampal-pituitary-adrenal axis, coupled with pro-inflammatory effects. They recognized that AngII does not readily cross the blood-brain barrier suggesting that it may be binding AT1 receptors located within circumventricular organs that are fenestrated permitting entry of larger molecules. The authors also indicated that peripherally infused AngII may activate AT1 receptors in the paraventricular nucleus of the hypothalamus. These issues must be resolved. One very important potential contributor to these depression-like behavioral responses is sustained elevations in blood pressure. Since blood pressure was not measured in this study, there is no way to determine whether systemic blood pressure reached hypertensive levels sufficient to cause lethargy in the treated mice. Even so these results are of potential importance and will require additional testing.
Identifying reliable biomarkers of depression has been challenging [292-294]. Many hypotheses have been posited to explain adulthood depression including alterations in glucocorticoid regulation and related stress hormones [295], insulin resistance [296], inflammatory chemokines and cytokines [297,298], and various trophic factors that are stimulated with injury, illness as well as other stressors [299]. Along these lines, accumulating evidence suggests that depression accompanying diabetes mellitus significantly increases pro-inflammatory mechanisms and a loss of hippocampal neuroplasticity [300-302]. The antidepressant medications presently available (5-hydroxytryptamine and norepinephrine-selective reuptake inhibitors) lack effectiveness in upwards of 50% of patients and typically require weeks of run-up treatment when effective [303].
Hippocampal and prefrontal cortex volume reductions
Post-mortem brain scans of depressed patients indicate significant reductions in the volume of limbic brain structures, most notably hippocampus and prefrontal cortex [304,305] two structures involved in memory and cognitive processing. Similar volume reductions (by MRI) have been measured in living MDD patients with severity depending on the progression of illness, duration and number of depressive episodes, and resistance to treatment [306]. Profound decreases in network connectivity have also been reported including decreases in intra- and inter-hemispheric functional connections. These results have been substantiated by a number of research groups [307-309] and have led to the notion that MDD should be categorized as a network dysfunctional disease [310]. Of particular importance, exposure to stress has been linked with neuronal atrophy and loss of glia in both structures [311,312]. Neurogenesis in the adult brain is known to occur in the sub-granular zone of the dentate gyrus of the hippocampus and subventricular zone of the lateral ventricles [313,314]. Neural stem cells in these structures are capable of dividing asymmetrically to form a daughter stem cell and a rapid multiplying progenitor cell. If appropriately stimulated these progenitor cells mature into neurons that integrate into functional neuronal networks [315,316]. Chronic stress-induced depression decreases neurogenesis; however, treatment with antidepressant drugs may reverse this process [311,317,318].
Neurotrophic growth factors
These observations point to the involvement of dysfunctional hippocampal plasticity in the neuropathology of depression, with particular focus on neurotrophic growth factors. The “neurotrophic hypothesis” of depression suggests that depression results from decreased neurotrophic growth factor activity causing atrophy of neurons in the hippocampus and prefrontal cortex, coupled with decreased neurogenesis and loss of glia. It has been hypothesized that treatment with antidepressant drugs interferes with, and/or blocks, neurotrophic factor deficits thus reversing atrophy [311-314]. The neurotrophic growth factors thus far linked with depression include Vascular Endothelial Growth Factor (VEGF), fibroblast growth factor-2, and insulin-like Growth Factor (IGF-1), with particular interest in Brain-Derived Neurotrophic Factor (BDNF) [319-323]. BDNF appears to be necessary for a positive response to treatment with antidepressant drugs [311,324]; however, preclinical results concerning the role of BDNF depletion in the etiology of depression are less consistent. BDNF-deletion mutant mice generally reveal normal behavior when tested for depression although conditional female mutant mice have been reported to show increased immobility during forced swim testing [325]. The use of RNA interference to knock down BDNF expression in hippocampal substructures results in depression as measured using forced swim and sucrose preference tasks [326]. Liu and colleagues [327] used a knock-in mouse prepared with human BDNF Val66met polymorphism in order to decrease trafficking of BDNF mRNA to dendrites. This resulted in reduced spine density and diameter and reduced synaptogenesis in the prefrontal cortex. Ketamine-induced synaptogenesis was impaired in these mice suggesting that synaptogenesis is dependent on dendritic translation/release of BDNF. In addition, the ketamine related antidepressant response seen in the forced-swim test was blocked. Human polymorphism in the BDNF gene appears to be carried by approximately 30% of the general population and is associated with mild cognitive deficits and depression. AngIV analogs acting at the Met receptor promotes synaptogenesis, neurogenesis and counters neuroinflammation. This approach may reduce the neuropathology and prevent neuron losses in the hippocampus and prefrontal cortex.
Neurodegenerative Diseases of the Eye
Approximately 3 million Americans have been diagnosed with glaucoma and 80 million worldwide. The overall number of patients is anticipated to reach 111 million by 2040 [328]. There are 11 million macular degeneration patients in the U.S. and this number is predicted to be 22 million by 2050. Worldwide there are about 196 million patients, predicted to approach 288 million by 2040 [328]. Diabetes is responsible for a significant number of new cases of retinopathy (12,000 to 24,000 cases) each year [329]. Currently there are 7.7 million Americans with diabetic retinopathy, a number expected to reach more than 14 million by 2050 [330]. Taken together these diseases represent the major reasons for blindness in the U.S. and around the world.
As previously discussed, several organs possess local reninangiotensin systems. This is true of the eye [331,332]. Major contributors to these diseases include increased Intra-Ocular Pressure (IOP), decreased retinal microvascular blood flow, tissue inflammation and oxidative stress [333]. The role of the local RAS of the eye will be discussed with respect to each of these biomarkers and related diseases.
Glaucoma
The RAS of the eye is a major regulatory factor in the normal maintenance of IOP. Continuous adjustment is necessary regarding Aqueous Humor (AH) flow. Optimal IOP is required in order to maintain the normal shape of the eye and in turn optical and refractory properties. AH is produced by the ciliary body [334] and exits the anterior chamber via the trabecular, uveoscleral and uveolymphatic pathways [335]. AH flow through the trabecular meshwork, the endothelial lining of Schlemm’s canal, and finally collateral channels and aqueous veins into the circulation [336-338]. This flow appears to be driven by the pressure gradient of the IOP [339-344]. The resistance against outflow yields an IOP of approximately 15 ± 5 mm Hg [334,345,346]. This value can vary depending on physical exercise [336,347-349], sleep, changes in posture [350], aging and disease [335].
The optic neuropathy caused by untreated glaucoma leads to death of retinal ganglion cells and neurons thus impacting vision [351,352]. This disease can be difficult to diagnose given that not every patient evidences an elevated IOP, and not all patients with an elevated IOP necessarily develop glaucoma [353]. However, at present of the known risk factors IOP is of major importance and pressure reduction procedures have been shown to slow disease progression [354-358]. These include topical medications, laser therapy and surgical intervention such as shunts designed to lower IOP by increasing AH outflow or procedures to decrease the formation of AH.
Igic and colleagues [359] were the first to measure the presence of ACE activity in retinal homogenates. Since that discovery all of the major components of the RAS have been isolated in the eye including AngII, Ang (1-7), ACE2, the AT1 and Mas receptors [331,360-362]. Given that the “blood-ocular” barrier discourages penetration by AngII, ACE and aldosterone it has been concluded that the eye possesses an intrinsic local RAS [363]. A number of investigators have proposed that this intrinsic system is important regarding the maintenance of IOP [332,364,365]. Related to this glaucoma patients, as well as animal models, placed on ACE inhibitors or ARBs evidence decreased IOP [366-372]. Glaucoma patients treated with the ARB losartan revealed significant drops in IOP via increased AH outflow regardless of whether they were initially hypertensive or not [368]. Further, topical application of Ang (1-7) also reduced IOP and this effect could be prevented by the Mas receptor antagonist A-779, suggesting that it was dependent on the Ang (1-7)/Mas receptor system [373]. There is also the claim that Ang (3-4) can inhibit ACE resulting in an increased level of AngI that is converted to Ang (1-7) by ACE2 and exerts a protective influence [374]. Some years ago, James Fitzsimons and others investigated the pressor potency of several centrally applied angiotensins [375-377]. They reported the greatest pressor activity to intracerebral injected AngII followed by AngI and AngIII (picomol range), with less activity induced by Ang (3-8), (4-8), (5-8), and (6-8) (nmol range). The C-terminal dipeptide AngII (7-8) and other dipeptides were inactive. Given that Ang (3-4) is a dipeptide with a peptide bond that is vulnerable to degradation in vivo, the half-life of Ang (3-4) is likely very short, casting doubt on the clinical usefulness of this peptide. However, additional testing is necessary to determine whether these results can be replicated.
Macular degeneration
Age-Related Macular Degeneration (AMD) is a neurodegenerative disease resulting in the progressive loss of photoreceptor retinal pigment Epithelial Complex (RPE) cells [378]. As this occurs there is the observable accumulation of extracellular material called drusen at the interface of the RPE and the inner collagenous zone of Bruch’s membrane. The detection of drusen within the macula of the eye is a definitive sign of AMD. The risk factors for AMD are many and include aging, genetics (offspring of AMD parents have a 3- to 6-fold increased risk as compared with the general population), smokers (greater than 40 years of smoking increases the likelihood by 2- to 4-fold), dietary intake of saturated fats, trans fats and omego-6 fatty acids [379], abdominal obesity also correlates, especially for men [380]. Chronic tissue inflammation is a recognized factor in AMD [381]. Although short term inflammation is very helpful to fight microbial infection and injuries, chronic inflammation can be very harmful, particularly as seen in neurodegenerative diseases. Finally, elevated oxidative stress and vascular insufficiency have been associated with inflammation, endothelial dysfunction and neuron degeneration in the retina [382].
There is a paucity of information concerning the potential role of the RAS in the etiology of AMD and yet there is overwhelming evidence that these local systems are instrumental in facilitating inflammation, increases in free radicals, coupled with vasoconstriction of local vessels [246]. As discussed, there are also angiotensin molecules capable of countering these deleterious factors including Ang (1-7) and small molecule AngIV analogs discussed in the next section.
Diabetic retinopathy
In general, the longer a patient has diabetes the greater the risk of developing Diabetic Retinopathy (DR) [383]. With non-proliferative DR the linings of retinal blood vessels are weakened resulting in microaneurysms. These bulges in the vessel wall often leak leading to swelling of the macula. With proliferative DR this condition advances to a critical point at which the retina is deprived of oxygen. In response new blood vessels form in the retina (angiogenesis) obstructing vision [384]. The tissue ischemia appears to trigger the production of growth factors such as VEGF. Diabetic animal models evidence increased retina levels of ACE, ACE2 and the AT1 receptor protein [385-387]. Since AngII has a mitogenic influence on retinal endothelial cells in the retinal microvasculature, it is a prime suspect in stimulating the up-regulation of VEGF [388,389]. In addition, it is known that AngII stimulates Reactive Oxygen Species (ROS) formation [390,391] and ROS promotes retinal damage in DR. Along these lines, diabetic animals treated with ACE inhibitors, or ARBs, evidenced reduced retinal microvascular damage, decreased vascular leakage, and reduced capillary formation and VEGF levels [392-395].
Human clinical trials to date have produced mixed results regarding the role of the RAS in the development of DR. One study reported that treatment with the ARB candesartan somewhat slowed retinopathy progression in Type 1 diabetic patients without hypertension [256,396-398]. A second study tested the combination of ACE inhibitors and a diuretic and found no impact on DR. It will be instructive to evaluate the efficacy of Ang (1-7), and the newly synthesized small molecule AngIV analogs, in the treatment of DR. These molecules should be tested by both oral and topical routes of administration.
Small Molecule Drug Development
Drug development targets
New targets must be considered in order to control the symptoms of neurodegenerative diseases and hopefully stop their progression. Clearly the RAS is a contributor and deserves particular attention. Basic characteristics of any drug candidate must include an extended half-life, the ability to protect vulnerable neurons in brain structures that mediate cognition, and the capacity to stop and perhaps reverse any damage. Additional specific criteria include the following: 1) the drug must penetrate the blood-brain-barrier in order to impact damaged brain structures. This is a major problem regarding most peptides and large proteins such as growth factors; 2) the halflife of the compound must be of sufficient duration to maintain a therapeutic level; 3) the avenue of drug delivery must be convenient for use by the patient and/or caregiver. This means oral, local application including cutaneous (patch) or subcutaneous (as with pen delivery) routes. Once the drug satisfies these challenges it would be desirable if it possessed the following neurological characteristics: 4) the capacity to encourage synaptogenesis, and promote stem cell proliferation, differentiation, and neurogenesis in impacted structures; 5) evidence neuroprotection especially against tissue ischemia, neuroinflammation and oxidative stress; 6) in the cases of dementia facilitate LTP, memory consolidation and retrieval, delay the onset of MCI and prevent concussion-induced encephalopathy. 7) With regard to the discussed eye diseases the drug candidate must protect the integrity of the retina and be delivered via convenient routes of administration.
Potential drug development approaches and targets
A first step in preventing the damage due to neurodegenerative diseases is to control hypertension with ARBs in order to block AT1 receptors or ACE inhibitors to reduce the synthesis of AngII. There is a need to collect additional information on the beneficial cognitive effects of these drugs. However, those normotensive individuals at risk to develop symptoms of MCI and dementias are likely not candidates for treatment with ACEi or ARBs. Also, at present there is minimal information concerning the potential effectiveness of these drugs to limit symptoms and no evidence that they prevent the onset of dementias.
Target: AngIII agonists acting at the AT2 receptor subtype
To date it is unclear whether the AT2 receptor is present in sufficient numbers in brain structures associated with cognitive functioning to warrant clinical testing in AD patients. This may change with additional research attention. An initial challenge concerns the design and synthesis of a non-peptidic AngIII agonist with an extended half-life that binds at the AT2 receptor subtype (Figure 3).
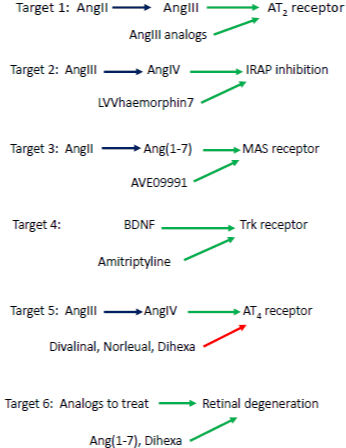
Figure 3: Suggested pharmacological targets to offset the deleterious
effects of an overactive Ang II/AT1 receptor subtype system. Target 1:
activation of the AT2 receptor by AngIII (and AngIII analogs) to initiate cellular
proliferation and differentiation accompanied by regenerative processes.
Target 2: AngIII conversion to AngIV (and AngIV analogs) acting at IRAP
results in IRAP inhibition thus resulting in the potentiation of several memory
enhancing peptides including AngIV, vasopressin, oxytocin, somatostatin
and cholecystokinin-8. Target 3: AngII conversion to Ang (1-7) that binds at
the MAS receptor. MAS receptor activation encourages the release of nitric
oxide thus promoting anti-thrombosis and facilitated Long-Term Potentiation
(LTP) resulting in enhanced memory processing. Target 4: Brain-Derived
Neurotrophic Factor (BDNF; and amitriptyline) binds at the Trk receptor
and increases trafficking of BDNF mRNA to dendrites. This appears to be
required for normal synaptogenesis. Target 5: AngIII is converted to AngIV
(also Dihexa) that acts as an agonist at the AT4 subtype resulting in facilitated
cerebral blood flow, increased neuroprotection, synaptogenesis and LTP,
thus promoting memory consolidation and retrieval. Small molecule AT4
receptor antagonists (Divalinal, Norleual) interfere with activation of the AT4
receptor subtype and may be particularly useful in decreasing the activation
of the Met receptor in solid tumors. Target 6: Ang (1-7) and small molecule
Ang (1-7) analogs, plus Dihexa, may be useful applied topically to the eye to
preserve the integrity of the retina by reducing oxidative stress, inflammation,
facilitating blood flow and thus decreasing the accumulation of extracellular
material called drusen.
Target: IRAP inhibitors
IRAP inhibitors (e.g. LVVhaemorphin7) have shown preclinical promise in enhancing memory on several tasks used to evaluate the performance of animal models [152,399]. Several specific inhibitors to IRAP have been developed that enhance spatial memory and fear avoidance in animal models [152,400]. Anderson and Hallberg [65] have been particularly successful in designing and synthesizing a number of IRAP inhibitors. Also, IRAP knockout mice have been shown to suffer significantly reduced cerebral infarct volume 24 hours following a 2-hour transient cerebral artery occlusion as compared with wild type mice [401]. These results were attributed to an increase in compensatory cerebral blood flow during the occlusion process. The authors suggest that IRAP may be an important target regarding the treatment of ischemic stroke as well as AD.
Target ang (1-7) analogs acting at the MAS receptor
A promising approach particularly regarding local ocular application concerns increased activation of the Ang (1-7)/Mas receptor system. Ang (1-7) has been shown to stimulate the release of Nitric Oxide (NO) from vascular endothelial and smooth muscle cells thus opposing AngII and vasopressin-induced vasoconstriction [402,403]. This peptide also protects cardiac and endothelium functioning as well as coronary perfusion as demonstrated in heart failure models [404]. It is of interest that Ang (1-7) has been shown to facilitate baroreceptor reflex sensitivity and modulate circadian rhythm influences on heart rate and blood pressure [405,406]. It is well established that AngII promotes thrombosis primarily via expression of Plasminogen Activator Inhibitor 1 (PAI-1) [407,408]. Kucharewicz and colleagues [409,410] have reported that Ang (1-7) functions as an antithrombotic agent when administered to renal hypertensive rats that served as a venous thrombosis model. A major first step toward the use of this peptide to offset AngII’s influence is the development of a non-peptidic Ang (1-7) analog, AVE09991 [411]. It would be very interesting to see the results of clinical trials designed to evaluate the efficacy of local application of this small molecule to glaucoma, macular degeneration and diabetic neuropathy patients.
Target: Neurotrophic analogs
There are few neurotransmitter, neuromodulatory or growth factor systems capable of satisfying the above listed drug development criteria and preventing dementia-associated dysfunctions. However, as earlier suggested neurotrophic agents possess characteristics that make them excellent candidates [13,76,80,246,412-414]. There are several neurotrophins capable of stimulating synaptogenesis, stem cell differentiation, neurogenesis, and neuroprotection against a wide range of cellular insults by facilitating anti-inflammatory and anti-apoptotic neuronal effects. These include NGF, BDNF, neurotrophin-3 and neurotrophin-4/5 [415-417]. Of these, BDNF has received considerable attention regarding depression and stress [418] and AD [176,412,416,419]. However, neurotrophins have had little success in clinical trials directed at neurodegenerative diseases due to their poor pharmokinetic profile and large molecular weight that significantly impedes penetration of the BBB [420,421]. Jang and colleagues [421,422] have reported that the small molecule antidepressant drug amitriptyline is capable of binding to the Trk receptor, induce receptor dimerization, and autophosphorylation. As previously mentioned receptor, dimerization is a prerequisite to activation of neurotrophins and downstream signaling. Thus, the use of amitriptyline may serve as a “short-cut” past BDNF to receptor activation and have a positive impact against AD. However, at the present time this drug is being tested in clinical trials conducted with depressed and chronic pain patients, but not AD patients [423].
Target: AngIV analogs acting at the AT4 receptor subtype
There is now substantial evidence that the brain AngIV/ AT4 receptor system is critically involved in memory formation and may overcome the memory inhibiting influences of AngII [54,55,87,131,424]. However, endogenous AngIV has a short halflife and thus appears to be over powered by AngII levels. In an effort to develop an AngIV, analog members of our laboratory initially synthesized a number of AngIV-based compounds possessing extended half-lives [425,426]. This resulted in the development of two potent receptor antagonists, divalinal-AngIV and norleual- AngIV [72,75,427-429], and one promising agonist, Nle1-AngIV. We determined that the memory facilitating effects of Nle1-AngIV derived from its N-terminal region given that fragments as small as tetra- and tripeptides retained the ability to overcome scopolamineinduced amnesia in animal models [73,87]. Further, Nle1-AngIV, as well as these shorter fragments, augmented hippocampal synaptic connectivity via the formation of new synapses [73]. Functionality of these synapses was established via evidence of analog-induced spinogenesis and the colocalization of synaptic markers in newly formed dendritic spines, which were coupled with enhanced miniature excitatory postsynaptic currents. These results encouraged the possibility that a clinically useful non-peptidic small molecule could be designed possessing increased metabolic stability with an extended half-life, and BBB penetrability offering facilitated cognitive functioning. Subsequent design and synthesis efforts yielded a small molecule with increased hydrophobicity, decreased hydrogen bonding potential, and significantly increased metabolic stability, dihexa. This compound induces spinogenesis/synaptogenesis at picomolar concentrations, is slowly cleared from the blood (plasma stability t1/2 = 5-6 hours) and can be delivered via parenteral routes of administration [76]. Dihexa binds with high affinity to HGF and stimulates dimerization, a prerequisite to binding at the Met receptor, and it induces Met phosphorylation in the presence of subthreshold levels of HGF. It also stimulates hippocampal spinogenesis and synaptogenesis equivalent with HGF [73,80]. Treatment with an HGF antagonist, “hinge”, as well as a short hairpin RNA directed at Met, significantly inhibited these actions suggesting that these effects are due to specific activation of the Met receptor. Further, dihexa penetrates the BBB in sufficient quantity to facilitate memory consolidation and retrieval in the scopolamine-induced amnesic rat model of AD, as well as in aged rats employing the Morris water maze task of spatial memory [54].
Target: Topically applied drugs to treat retinal degeneration
Given results, indicating that ACE inhibitors and ARBs reduce ocular pressure the topical application of these drugs to glaucoma patients is worth evaluating. A clinical trial utilizing topically applied Ang (1-7) in glaucoma patients while monitoring ocular pressure and progression of retinal damage should be conducted. Regarding macular degeneration and diabetic retinopathy patients, topically applied Ang (1-7) and dihexa should be investigated with ongoing measurements of the same dependent measures.
Conclusion
Progress is being made concerning early detection of MCI. The development of new efficacious drugs to delay, and hopefully prevent, the onset of dementia symptoms must catch up with these efforts. This will require a shift in our thinking. This shift is supported by the following observations from past findings: 1) β-amyloid-induced plaques and neurofibrillary tangles define AD but may not cause it. These cellular markers are likely consequential to other deleterious dysfunctions. 2) Efforts to develop drugs to rid neurons of amyloid plaque buildup have generally failed to improve cognitive processing. It is likely that current efforts to prevent neurofibrillary tangles will also fall short with regard to improved memory functioning. 3) Alzheimer’s disease has a multitude of potential causes. These include, but are not limited to, genetic predisposition, neuroinflammation, head trauma, untreated hypertension, diabetes, Parkinson’s disease, infection and normal aging. It may be necessary to attack each of these likely causes with separate drug development programs. 4) Presently available drugs do not promote synaptogenesis of existing neurons. The loss of synaptic connections discourages neurogenesis and is a major cause of neuronal apoptosis; 5) Current research must encourage efforts to develop drugs that penetrate the BBB, facilitate cognitive processing, and protect against the loss of synapses and neurons. 6) Neurotrophic agents offer the ability to facilitate synaptogenesis, neurogenesis and neuroprotection thus greater research attention must be directed toward creating small molecule analogs designed to penetrate the BBB and activate these brain systems. 7) It is likely that a successful approach to treating AD will require several different “Multiple Target-Directed Ligands” (MTDLs). Neurotrophic small molecule analogs may be useful in configuring such a strategy. 8) New drugs must be developed to treat glaucoma, macular degeneration, and diabetic retinopathy. With each disease, a major consequence is progressive retinal damage. The research presently summarized suggests that compounds designed to reduce the influence of the local AngII/AT1 receptor system should be effective in controlling retinal damage in patients afflicted with these dysfunctions. Small molecule RAS related drugs designed to function as AT1 receptor antagonists, followed by AngIV small molecule analogs that pass the blood-retina barrier, act to reduce oxidative stress, facilitate blood flow and stem neurodegeneration, are promising candidates as ocular treatments.
Declaration
Competing interest: Drs. Wright and Harding are the cofounders of M3 Biotechnology, Inc. (now Athira Pharma, Inc.) and hold stock in this company. No funds from this company were used to conduct the animal research presented in this manuscript or in its preparation. The authors are no longer actively involved in this company and have made every effort to objectively discuss the theories, findings and conclusions without bias regarding these topics.
Acknowledgements: All experiments utilizing animal models conducted in our laboratory and referred to in this review adhered to the Guidelines for the Care and Use of Laboratory Animals as required by the National Institutes of Health (NIH Publication No. 80-23), and the protocols were approved by the Washington State University Institutional Animal Care and Use Committee. The preparation of this paper was supported by the Michael J Fox Foundation, and the Alzheimer’s Drug Discovery Foundation.
We have been privileged to benefit from conversations with, and conference presentations and papers by, insightful researchers interested in unraveling the mysteries and components of the reninangiotensin system. These scientists included: Professors Merlin Bumpus, Arthur C Guyton, William Ganong, James Fitzsimons, Detlef Ganten, Dominic Felix, James Fitzsimons, Mike Peach, Michael Brody, Alan Epstein, Ian Phillips, Fred Mendelsohn, Kim Johnson, Mark de Gasparo, Shigi Mitzutani and many others. Over the years, we have maintained a strong interest concerning the ever-increasing importance and complexity of the RAS, and anticipate this system to disclose many additional important functions and surprises in the future. We urge young neuroscientists to continue in the footsteps of these gifted researchers bringing enthusiasm and energy to the important task of discovering new functions of the renin-angiotensin system.
References
- Fyhrquist F, Saijonmaa O. Renin-angiotensin system revisited. J Internal Med. 2008; 264: 224-236.
- Braun-Menendez E, Fasiolo JC, Leioir LF, Munoz JM. The substance causing renal hypertension. J Physiol. 1940; 98: 283-298.
- Page IH and Helmer OM. A crystalline pressor substance (angiotonin) resulting from the action between renin and renin-activator. J Exp Med. 1940; 71: 29-42.
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000; 52: 415-472.
- Bumpus FM, Schwarz H, Page IH. Synthesis and pharmacology of the octapeptide angiotonin. Science. 1957; 125: 886-887.
- Bumpus FM, Schwarz H, Page IH. Synthesis and properties of angiotensin. Circulation. 1958; 17: 664-667.
- Skeggs LT, Kahn JR, Lentz KE, Shumway NP. The preparation, purification and amino acid sequence of polypeptide renin substrate. J Exp Med. 1957; 106: 439-453.
- Dupont AG, Brouwers S. Brain angiotensin peptides regulate sympathetic tone and blood pressure. J Hypertens. 2010; 28: 1599-1610.
- Rompe F, Unger T, Steckelings UM. The angiotensin AT2 receptor in inflammation. Drug News Perspect. 2010; 23: 104-111.
- Sandberg K, Ji H, Clark AJ, Shapira H, Catt KJ. Cloning and expression of a novel angiotensin II receptor subtype. J Biol Chem. 1992; 267: 9455-9458.
- Inagami T, Iwai N, Sasaki K, Guo DF, Furuta H, Yamano Y et al. Angiotensin II receptors: Cloning and regulation. Arzneimittelforschung. 1993; 43: 226- 228.
- Wright JW, Kawas LH, Harding JW. A role for the brain RAS in Alzheimer’s and Parkinson’s diseases. Front Endocrin. 2013; 4: 158.
- Wright JW, Kawas LH, Harding JW. The development of small molecule angiotensin IV analogs to treat Alzheimer’s and Parkinson’s diseases. Prog Neurobiol. 2015; 125: 26-46.
- Sandberg K, Ji H, Catt KJ. Regulation of angiotensin II receptors in rat brain during dietary sodium changes. Hypertension. 1994; 23: I-137-I-141.
- Unger T, Chung O, Csikos T, Culman J, Gallinat,S, Gohlke P, et al. Angiotensin receptors. J Hypertens. 1996; 14: S95-S103.
- Allen AM, Moeller I, Jenkings TA, Zhuo J, Aldred GP, Chai SY, et al. Angiotensin receptors in the nervous system. Brain Res Bull. 1998; 47: 17- 28.
- Phillips MI, de Oliveira EM. Brain renin angiotensin in disease. J Mol Med. 2008; 86: 715-722.
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010; 140: 918-934.
- Ownby RL. Neuroinflammation and cognitive aging. Curr Psychiatry Rep. 2010; 12: 39-45.
- Ohshima K, Mogi M, Horiuchi M. Therapeutic approach for neuronal disease by regulating renin-angiotensin system. Curr Hypert Rev. 2013; 9: 99-107.
- Di Marco LY, Venneri A, Farkas E, Evans PC, Marzo A. Vascular dysfunction in the pathogenesis of Alzheimer’s disease - A review of endotheliummediated mechanisms and ensuing vicious circles. J Alzheimers Dis. 2015; 82: 593-606.
- Johnston CI. Biochemistry and pharmacology of the renin-angiotensin system. Drugs. 1990; 39: 21-31.
- Speth RC, Karamyan VT. The significance of brain aminopeptidases in the regulation of the actions of angiotensin peptides in the brain. Heart Fail Rev. 2008; 13: 299-309.
- Bader M. Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Annu Rev Pharmacol Toxicol. 2010; 50: 439-465.
- Cosarderelioglu C, Nidadavolu LS, George CH, Oh ES, Bennett DA, Watston JD, et al. Brain renin-angiotensin system at the intersect of physical and cognitive frailty. Front Neurosci. 2020; 14: 586314.
- Rich DH, Moon BJ, Harbeson S. Inhibition of aminopeptidases by amastatin and bestatin derivatives, effect of inhibitor structure on slow-binding processes. J Med Chem. 1984; 27: 417-422.
- Wilk S, Healy D. Glutamyl aminopeptidase (aminopeptidase A), the BP- 1/6C3 antigen. Adv Neuroimmunol. 1993; 3: 195-207.
- Chauvel EN, Llorens-Cortes C, Coric P, Wilk S, Roques BP, Fournie-Zaluski MC. Differential inhibition of aminopeptidase A and aminopeptidase N by new -amino thiols. J Med Chem. 1994; 37: 2950-2957.
- Wright JW, Bechtholt AJ, Chambers SL, Harding JW. Angiotensin III and IV activation of the brain AT1 receptor subtype in cardiovascular function. Peptides. 1996; 17: 1365-1371.
- Ferrario CM, Chappell MD. Novel angiotensin peptides. Cell Mol Life Sci. 2004; 61: 2720-2727.
- Ohishi M, Yamamoto K, Rakugi H. Angiotensin (1-7) and other angiotensin peptides. Curr Pharm Des. 2013; 19: 3060-3064.
- Vauquelin G, Michotte Y, Smolders I, Sarre S, Ebinger G, Dupont A, et al. Cellular targets for angiotensin II fragments: pharmacological and molecular evidence. J Renin Angiotensin Aldosterone Syst. 2002; 3: 195-204.
- Sayeski PP, Ali MS, Semeniuk DJ, Doan TN, Bernstein KE. Angiotensin II signal transduction pathways. Regul Pept. 1998; 78: 19-29.
- Dinh DT, Grauman AG, Johnston CI, Fabiani ME. Angiotensin receptors: distribution, signaling and function. Clin Sci. 2001; 100: 481-492.
- Wright JW, Harding JW. Important roles for angiotensin III and IV in the brain renin-angiotensin system. Brain Res Rev. 1997; 25: 96-124.
- Unger T, Badoer E, Ganten D, Lang RE, Rettig R. Brain angiotensin: Pathways and pharmacology. Circulation. 1988; 77: 140-154.
- Phillips MI, Sumners C. Angiotensin II in central nervous system physiology. Regul Pept. 1998; 78: 1-11.
- Culman J, Blume A, Gohlke P, Unger T. The renin-angiotensin system in the brain: Possible therapeutic implications for AT1-receptor blockers. J Hum Hypertens. 2002; 16: S64-S70.
- Bottari SP, Raylor V, King IN, Bogdal S, Whitebread S, DeGasparo M. Angiotensin II AT2 receptors do not interact with guanine nucleotide binding proteins. Eur J Pharmacol. 1991; 207: 157-163.
- Kambayashi Y, Bardham S, Takahashi K, Tsuzuki S, Inui H, Hamakubo T, et al. Molecular cloning of a novel angiotensin II receptor isoform involved in phosphotyrosine phosphatase inhibition. J Biol Chem. 1993; 268: 24543- 24546.
- Nakajima M, Mukoyama M, Pratt RE, Horiuchi M, Dzau VJ. Cloning of cDNA and analysis of the gene for mouse angiotensin II type 2 receptor. Biochem Biophys Res Commun. 1993; 197: 393-399.
- Wright JW, Yamamoto BJ, Harding JW. Angiotensin receptor subtype mediated physiologies and behaviors: New discoveries and clinical targets. Prog Neurobiol. 2008; 84: 157-181.
- Rosenstiel P, Gallinat S, Arit A, Unger T, Sievers J, Lucius R. Angiotensin AT2 receptor ligands: Do they have potential as future treatments for neurological disease? CNS Drugs. 2002; 16: 145-153.
- Rodriguez-Pallares J, Quiroz CR, Parga JA, Guerra MJ, Labandeira-Garcia JL. Angiotensin II increases differentiation of dompaminergic neurons from mesencephalic precursors via angiotensin type 2 receptors. Eur J Neurosci. 2004; 20: 1489-1498.
- Chabrashvili T, Kitilyakara C, Blau J, Karber A, Asiam S, Welch WJ, et al. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol. 2003; 285: R117-R124.
- Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE. AT2 receptors: Functional relevance in cardiovascular disease. Pharmacol Ther. 2008; 120: 292-316.
- Jarvis MF, Gessner GW, Ly CG. The angiotensin hexapeptide 3-8 fragment potently inhibits [125I] angiotensin II binding to non-AT1 or -AT2 recognition sites in bovine adrenal cortex. Eur J Pharmacol. 1992; 219: 319-322.
- Swanson GN, Hanesworth JM, Sardinia MF, Coleman JK, Wright JW, Hall KL, et al. Discovery of a distinct binding site for angiotensin II (3-8), a putative angiotensin IV receptor. Regul Pept. 1992; 40: 409-419.
- Bernier SG, Fournier A, Guillemette G. A specific binding site recognizing a fragment of angiotensin II in bovine adrenal cortex membranes. Eur J Pharmacol. 1994; 271: 55-63.
- Harding JW, Cook VI, Miller-Wing AV, Hanesworth JM, Sardinia MF, Hall KL, et al. Identification of an AII (3-8) [AIV] binding site in guinea pig hippocampus. Brain Res. 1992; 583: 340-343.
- Wright JW, Harding JW. The brain angiotensin system and extracellular matrix molecules in neural plasticity, learning, and memory. Prog Neurobiol. 2004; 72: 263-293.
- Wright JW, Harding JW. Brain renin-angiotensin: A new look at an old system. Prog Neurobiol. 2011; 95: 49-67.
- Wright JW, Stubley L, Pederson ES, Kramár EA, Hanesworth JM, Harding JW. Contributions of the brain angiotensin IV-AT4 receptor subtype system to spatial learning. J Neurosci. 1999; 19: 3952-3961.
- Wright JW, Harding JW. The brain hepatocyte growth factor/c-Met receptor system: A new target for the treatment of Alzheimer’s disease. J Alzheimers Dis. 2015; 45: 985-1000.
- Braszko JJ, Kupryszewski G, Witczuk B, Wisniewski K. Angiotensin II-(3-8)- hexapeptide affects motor activity, performance of passive avoidance and conditioned avoidance responses in rats. Neuroscience. 1988; 27: 777-783.
- Braszko JJ, Wlasienko J, Koziolkiewicz W, Janecka A, Wisniewski K. The 3-7 fragment of angiotensin II is probably responsible for its psychoactive properties. Brain Res. 1991; 542: 49-54.
- Braszko JJ. Indispensable role of the voltage-gated calcium channels in the precognitive effects of angiotensin IV. Brain Res Bull. 2017; 130: 118-124.
- Wright JW, Miller-Wing AV, Shaffer MJ, Higginson C, Wright DE, Hanesworth JM, et al. Angiotensin II(3-8) (ANG IV) hippocampal binding: potential role in the facilitation of memory. Brain Res Bull. 1993; 32: 497-502.
- Kramár EA, Harding JW, Wright JW. Angiotensin II- and IV-induced changes in cerebral blood flow. Roles of AT1, AT2, and AT4 receptor subtypes. Regul Pept. 1997; 68: 131-138.
- Naveri L, Stromberg C, Saavedra JM. Angiotensin IV reverses the acute cerebral blood flow reduction after experimental subarachnoid hemorrhage in the rat. J Cereb Blood Flow Metab. 1994; 14: 1096-1099.
- Dalmay F, Pesteil F, Allard J, Nisse-Durgeat S, Fernandez L, Fournier A. Angiotensin IV decreases acute stroke mortality in the gerbil. Hypertension. 2001; 14: 56A.
- Faure S, Chapot R, Tallet D, Javellaud J, Achard JM, Oudart N. Cerebroprotective effect of angiotensin IV in experimental ischemic stroke in the rat mediated by AT(4) receptors. J Physiol Pharmacol. 2006; 57: 329- 342.
- Albiston AL, McDowall SG, Matsacos D, Sim P, Clune E, Mustafa T, et al. Evidence that the angiotensin IV (AT4) receptor is the enzyme insulin regulated aminopeptidase. J Biol Chem. 2001; 276: 48263-48266.
- Albiston AL, Mustafa T, McDowall SG, Mendelsohn FA, Lee J, Chai SY. AT(4) receptor is insulin-regulated aminopeptidase: Potential mechanisms of memory enhancement. Trends Endocrinol Metab. 2003; 14: 72-77.
- Andersson H, Hallberg M. Discovery of inhibitors of insulin-regulated aminopeptidase as cognitive enhancers. Int J Hypertens. 2012: 12.
- Kandror KV, Pilch PF. Gp160, a tissue-specific marker for insulin-activated glucose transport. Proc Natl Acad Sci USA. 1994; 91: 8017-8021.
- Keller SR, Scott HM, Mastick CC, Aebersold R, Lienhard GE. Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles. J Biol Chem. 1995; 270: 23612-23618.
- Albiston AL, Fernando RN, Yeatman HR, Burns P, Ng I, Daswani D, et al. Gene knock-out of insulin-regulated aminopeptidase: Loss of the specific binding site for angiotensin IV and age-related deficit in spatial memory. Neurobiol Learn Mem. 2010; 93: 19-30.
- De Bundel D, Smolders I, Vanderheyden P, Michotte Y. AngII and AngIV: Unraveling the mechanism of action on synaptic plasticity, memory, and epilepsy. CNS Neurosci Ther. 2008; 14: 315-339.
- Lew RA, Mustafa T, Ye S, McDowall SG, Chai SY, Albiston AL. Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP). J Neurochem. 2003; 86: 344-350.
- Yeatman HR, Albiston AL, Burns P, Chai SY. Forebrain neurone-specific deletion of insulin-regulated aminopeptidase causes age related deficits in memory. Neurobiol Learn Mem. 2016; 136: 174-182.
- Yamamoto BJ, Elias PD, Masino JA, Hudson BD, McCoy AT, Anderson ZJ, et al. The angiotensin IV analog Nle-Tyr-Leu-ψ-(CH2-NH2)3-4-His-Pro-Phe (Norleual) can act as a hepatocyte growth factor/c-Met inhibitor. J Pharmacol Exp Ther. 2010; 333: 161-173.
- Benoist CC, Wright JW, Zhu M, Appleyard SM, Wayman GA, Harding JW. Facilitation of hippocampal synaptogenesis and spatial memory by C-terminal truncated Nle1-angiotensin IV analogs. J Pharmacol Exp Ther. 2011; 339: 35-44.
- Wright JW, Harding JW. Importance of the brain angiotensin system in Parkinson’s disease. Parkinson’s Dis. 2012; 2012: 860923.
- Kawas LH, McCoy AT, Yamamoto BJ, Wright JW, Harding JW. Development of angiotensin IV analogs as hepatocyte growth factor/Met modifiers. J Pharmacol Exp Ther. 2012; 340: 539-548.
- McCoy AT, Benoist CC, Wright JW, Kawas LH, Bule-Ghogare JM, Zhu M, et al. Evaluation of metabolically stabilized angiotensin IV analogs as procognitive/antidementia agents. J Pharmacol Exp Ther. 2013; 344: 141- 154.
- Kawas LH, Yamamoto BJ, Wright JW, Harding JW. Mimics of the dimerization domain of hepatocyte growth factor exhibit anti-Met and anticancer activity. J Pharmacol Exp Ther. 2011; 339: 509-518.
- Gherardi E, Sandin S, Petoukhov MV, Finch J, Youles ME, Ofverstedt LG, et al. Structural basis of hepatocyte growth factor/scatter factor and MET signaling. Proc Natl Acad Sci USA. 2006; 103: 4046-4051.
- Youles M, Holmes O, Petoukhov MV, Nessen MA, Stivala S, Svergun DI, et al. Engineering the NK1 fragment of hepatocyte growth factor/scatter factor as a MET receptor antagonist. J Mol Biol. 2008; 377: 616-622.
- Benoist CC, Kawas LH, Zhu M, Tyson KA, Stillmaker L, Appleyard M, et al. The pro-cognitive and synaptogenic effects of angiotensin IV-derived peptides are dependent on activation of the hepatocyte growth factor/c-Met system. J Pharmacol Exp Ther. 2014; 351: 390-402.
- Albiston AL, Pederson ES, Burns P, Purcell B, Wright JW, Harding JW, et al. Attenuation of scopolamine-induced learning deficits by LVV-hemorphin-7 in rats in the passive avoidance and water maze paradigms. Behav Brain Res. 2004; 154: 239-243.
- Hellner K, Walther T, Schubert M, Albrecht D. Angiotensin-(1-7) enhances LTP in the hippocampus through the G protein-coupled receptor Mas. Mol Cell Neurosci. 2005; 29: 427-435.
- Passos-Silva DG, Verano-Braga T, Santos RA. Angiotensin-(1-7): Beyond the cardio-renal actions. Clin Sci (Lond). 2013; 124: 443-456.
- Simoes e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. 2013; 169: 477-492.
- Freund M, Walther T, von Bohlen und Halbach O. Immunohistochemical localization of the angiotensin-(1-7) receptor Mas in the murine forebrain. Cell Tissue Res. 2012; 348: 29-35.
- Dasgupta C, Zhang L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov Today. 2011; 16: 22-34.
- Wright JW, Harding JW. The brain angiotensin IV/AT4 receptor system as a new target for the treatment of Alzheimer’s disease. Drug Dev Res. 2009; 70: 472-480.
- Ganten D, Boucher R, Genest J. Renin activity in brain tissue of puppies and adult dogs. Brain Res. 1971; 33: 557-559.
- Ganten D, Marquez-Julio A, Granger P, Hayduk K, Karsunky KP, Boucher R. Renin in dog brain. Am J Physiol. 1971; 221: 1733-1737.
- Ganten D, Hermann K, Bayer D, Unger T Lang RE. Angiotensin synthesis in the brain and increased turnover in hypertensive rats. Science. 1983; 221: 869-871.
- Campbell DJ, Habener JF. Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat. J Clin Invest. 1986; 78: 31-39.
- Deschepper DF, Bouhnik J, Ganong WF. Colocalization of angiotensinogen and glial fibrillary acidic protein in astrocytes in rat brain. Bran Res. 1986; 374: 195-198.
- Leung PS. Novel roles of a local angiotensin-generating system in the carotid body. J Physiol. 2006; 575: 4.
- Leung PS. Local RAS. The Renin-Angiotensin System. Curr Res Prog Pancreas. 2010; 690: 69-87.
- Husain A, Bumpus FM, De Silva P, Speth RC. Localization of angiotensin II receptors in ovarian follicles and the identification of angiotensin II in rat ovaries. Proc Natl Acad Sci USA. 1987; 84: 2489-2493.
- Wright JW, Harding JW. Regulatory role of brain angiotensins in the control of physiological and behavioral responses. Brain Res Rev. 1992; 17: 227- 262.
- Mangiapane ML, Simpson JB. Subfornical organ: Forebrain site of pressor and dipsogenic action of angiotensin II. Am J Physiol. 1980; 239: R382-R389.
- Brody MJ. Central nervous system mechanisms of arterial pressure regulation. Fed Proc. 1986; 45: 2700-2706.
- Phillips MI. Functions of angiotensin in the central nervous system. Annu Rev Physiol. 1987; 49: 413-435.
- Muratami H. Brain angiotensin and circulatory control. Clin Exp Pharmacol Physiol. 1996; 23: 458-464.
- Unger T, Becker H, Petty M, Demmert C, Schneider B, Ganten D, et al. Differential effects of central angiotensin II and substance P on sympathetic nerve activity in conscious rats. Circ Res. 1985; 56: 563-575.
- Head GA. Role of AT1 receptors in the central control of sympathetic vasomotor function. Clin Exp Pharmacol Physiol. 1996; 23: S93-S98.
- Allen AM, Zhuo J, Mendelsohn FA. AT1-receptors in the central nervous system. J Renin Angiotensin Aldosterone Syst. 2001; 2: S95-S101.
- Khosla MC, Leese RA, Maloy WL, Ferreira AT, Smeby RR, Bumpus FM. Synthesis of some analogs of angiotensin II as specific antagonists of the parent hormone. J Med Chem. 1972; 15: 792-795.
- Brunner HR, Gavras H, Laragh JH, Keenan R. Angiotensin II blockade in man by SAR ALA angiotensin II for understanding and treatment of high blood pressure. Lancet. 1973; 2: 1045-1048.
- Haber E. The role of renin in normal and pathological cardiovascular homeostasis. Circulation. 1976; 54: 849-861.
- Streeten DHP, Anderson GH Jr, Dalakos TG. Angiotensin blockade: Its clinical significance. Amer J Med. 1976; 60: 817-824.
- Wexler RR, Greenlee WJ, Irvin JD, Goldberg MR, Prendergast K, Smith RD, et al. Nonpeptide angiotensin II receptor antagonists: The next generation in antihypertensive therapy. J Med Chem. 1996; 39: 625-656.
- Kjeldsen SE, Stlhammar J, Hasvold P, Bodegard J, Olsson U, Russell D. Effects of losartan vs candesartan in reducing cardiovascular events in the primary treatment of hypertension. J Hum Hypertens. 2010; 24: 263-273.
- Eklind-Cervenka M, Benson L, Dahlstrom U, Edner M, Rosenqvist M, Lund LH. Association of candesartan vs losartan with all-cause mortality in patients with heart failure. JAMA. 2011; 305: 175-182.
- White WB, Weber MA, Sica D, Bakris GL, Perez A, Cao C, et al. Effects of the angiotensin receptor blocker azilsartan medoxomil versus olmesartan and valsartan on ambulatory and clinic blood pressure in patients with stages 1 and 2 hypertension. Hypertension. 2011; 57: 413-420.
- Basso N, Paglia N, Stella I. Protective effect of the inhibition of the reninangiotensin system on aging. Regul Pept. 2005; 128: 247-252.
- Hajjar IM, Keown M, Frost B. Antihypertensive agents for aging patients who are at risk for cognitive dysfunction. Curr Hypert Rep. 2005; 7: 466-473.
- Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, et al. Antihypertensive medication use and incident Alzheimer disease: The Cache County study. Arch Neurol. 2006; 63: 686-692.
- Mogi M, Horiuchi M. Effects of angiotensin II receptor blockers on dementia. Hypertens Res. 2009; 32: 738-740.
- Ondetti MA, Rubin B, Cushman DW. Design of specific inhibitors of angiotensin converting enzyme: New class of orally active antihypertensive agents. Science. 1977; 196: 441-444.
- Todd PA, Heel RC. Enalapril. A review of its pharmacodynamics and pharmacokinetic properties, and therapeutic use in hypertension and congestive heart failure. Drugs. 1986; 31: 198-248.
- Bauer JH. Angiotensin converting enzyme inhibitors. Am J Hypertens. 1990; 3: 331-337.
- Ellul J, Archer N, Foy CM, Poppe M, Boothby H, Nicholas H, et al. The effects of commonly prescribed drugs in patients with Alzheimer’s disease on the rate of deterioration. J Neurol Neurosurg Psychiatry. 2006; 78: 233- 239.
- Croog SH, Levine S, Testa MA, Brown B, Bulpitt CJ, Jenkins CD, et al. The effects of antihypertensive therapy on the quality of life. New Engl J Med. 1986; 314: 1657-1674.
- Amenta F, Di Tullio MA, Tomassoni D. The cholinergic approach for the treatment of vascular dementia: evidence from pre-clinical and clinical studies. Clin Exp Hypertens. 2002; 24: 697-713.
- Rozzini L, Chilovi BV, Bertoletti E, Conti M, Del Rio L, Trabucchi M, et al. Angiotensin converting enzyme (ACE) inhibitors modulate the rate of progression of amnestic mild cognitive impairment. Internat J Geriat Psychiat. 2006; 21: 550-555.
- Kehoe PG, Wilcock GK. Is inhibition of the renin-angiotensin system a new treatment option for Alzheimer’s disease? Lancet Neurol. 2007; 6: 373-378.
- Hajjar IM, Keown M, Lewis P, Almor A. Angiotensin converting enzyme inhibitors and cognitive and functional decline in patients with Alzheimer’s disease: An observational study. Am J Alzheimers Dis Other Demen. 2008; 23: 77-83.
- Soto ME, van Kan GA, Nourhashemi F, Gillette-Guyonnet S, Cesari M, Cantet C, et al. Angiotensin-converting enzyme inhibitors and Alzheimer’s disease progression in older adults: Results from the Reseau sur la Maladie d-Alzheimer Francais cohort. J Am Geriatr Soc. 2013; 61: 1482-1488.
- Sudilovsky A, Cutler NR, Sramek JJ, Wardle T, Veeroff AE, Mickelson W, et al. A pilot clinical trial of the angiotensin-converting enzyme inhibitor ceranapril in Alzheimer’s disease. Alzheimer Dis Assoc Disord. 1993; 7: 105-111.
- Khachaturian AS, Zandi PP, Lyketsos G, Hayden KM, Skoog I, Norton MC, et al. Antihypertensive medication use and incident Alzheimer disease: the Cache County study. Arch Neurol. 2006; 63: 686-692.
- Ashby EL, Kehoe PG. Current status of renin-aldosterone angiotensin system-targeting anti-hypertensive drugs as therapeutic options for Alzheimer’s disease. Expert Opin Investig Drugs. 2013; 22: 1229-1242.
- Mustafa T, Lee JH, Chai SY, Albiston AL, McDowall SG, Mendelsohn FA. Bioactive angiotensin peptides: Focus on angiotensin IV. J Renin Angiotensin Aldosterone Syst. 2001; 2: 205-210.
- Gard PR. Angiotensin as a target for the treatment of Alzheimer’s disease, anxiety and depression. Expert Opin Ther Targets. 2004; 8: 7-14.
- Gard PR. Cognitive-enhancing effects of angiotensin IV. BMC Neurosci. 2008; 9: S2-S15.
- Ohrui T, Matsui T, Yamaya M, Arai H, Ebihara S, Maruyama M, et al. Angiotensin-converting enzyme inhibitors and incidence of Alzheimer’s disease in Japan. J Am Geriatr Soc. 2004; 52: 649-650.
- Yamada K, Horita T, Takayama M, Rakahashi S, Takaba K, Nagata Y, et al. Effect of a centrally active angiotensin converting enzyme inhibitor, perindopril, on cognitive performance in chronic cerebral hypo-perfusion rats. Brain Res. 2011; 1421: 110-120.
- Handa RK. Angiotensin-(1-7) can interact with the rat proximal tubule AT(4) receptor system. Am J Physiol. 1999; 277: F75-83.
- Pederson ES, Krishnan R, Harding JW, Wright JW. A role for the angiotensin AT4 receptor subtype in overcoming scopolamine-induced spatial memory deficits. Regul Pept. 2001; 102: 147-156.
- Stubley-Weatherly L, Harding JW, Wright JW. Effects of discrete kainic acidinduced hippocampal lesions on spatial and contextual learning and memory in rats. Brain Res. 1996; 716: 29-38.
- McFall A, Nicklin SA, Work LM. The counter regulatory axis of the renin angiotensin system in the brain and ischaemic stroke: Insight from preclinical stroke studies and therapeutic potential. Cell Signal. 2020; 76: 109809.
- Wright JW, Clemens JA, Panetta JA, Smalstig EB, Weatherly A, Kramár EA, et al. Effects of LY231617 and angiotensin IV on ischemia-induced deficits in circular water maze and passive avoidance performance in rats. Brain Res. 1996; 717: 1-11.
- de la Torre JC. Alzheimer’s disease: How does it start? J Alzheimers Dis. 2002; 4: 497-512.
- Chai SY, Bastias MA, Clune EF, Matsacos DJ, Mustafa T, Lee JH, et al. Distribution of angiotensin IV binding sites (AT4 receptor) in the human forebrain, midbrain and pons as visualized by in vitro receptor autoradiography. J Chem Neuroanat. 2000; 20: 339-348.
- Yaari R, Corey-Bloom J. Alzheimer’s disease. Semin Neurol. 2007; 27: 32- 41.
- Honig LS, Boyd CD. Treatment of Alzheimer’s disease: Current management and experimental therapeutics. Curr Transl Geniatr Exp Gerontol Rep. 2013; 2: 174-181.
- Clark CM. Clinical manifestations and diagnostic evaluation of patients with Alzheimer’s disease. In: Clark CM and Trajanowski JQ (Eds.), Neurodegenerative dementias: Clinical features and pathological mechanisms. McGraw-Hill, New York, N.Y. 2000: 95-114.
- Brockmeyer R, Abdalla N, Kawas CH, Corrada MM. Forcasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimer’s and Dement. 2018; 14: 981-988.
- Suehs BT, Davis CD, Alvir J, van Ameronnen D, Pharmd NC, Joshi AV, et al. The clinical and economic burden of newly diagnosed Alzheimer’s disease in a medicare advantage population. Am J Alzheimers Dis Other Dement. 2013; 28: 384-392.
- Maiese K, Chong ZZ, Hou J, Shang YC. New strategies for Alzheimer’s disease and cognitive impairment. Oxid Med Cell Longev. 2009; 2: 279-289.
- Zec RF, Trivedi MA. The effects of estrogen replacement therapy on neuropsychological functioning in postmenopausal women with and without dementia: A critical and theoretical review. Neuropsychol Rev. 2002; 12: 65- 109.
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and meta-analysis. Alzheimers Dement. 2013; 9: 63-75.
- Albayram O, Herbert MK, Kondo A, Tsai CY, Baxley S, Lian X, et al. Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci. 2016; 6: 59.
- Mez J, Daneshyar DH, Kiernan PT, Abdolmohammadi B, Alarez VE, Huber BR. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. 2017; 318: 360-370.
- Randolph C, Karantzoulis S, Guskiewicz K. Prevalence and characterization of mild cognitive impairment in retired national football league players. J Int Neuropsychol Soc. 2013; 19: 873-880.
- Albiston AL, Diwakaria S, Fernando RN, Mountford SJ, Yeatman H, Morgan B, et al. Identification and development of specific inhibitors for insulinregulated aminopeptidase as a new class of cognitive enhancers. Br J Pharmacol. 2011; 164: 37-47.
- Briggs R, Kennelly SP, O’Neill DO. Drug treatments in Alzheimer’s disease. Clin Med. 2016; 16: 247-253.
- Douchamps V, Mathis C. A second wind for the cholinergic system in Alzheimer’s therapy. Behav Pharmacol. 2017; 28: 112-123.
- Melnikova I. Therapies for Alzheimer’s disease. Nat Rev Drug Discov. 2007; 6: 341-342.
- Thomas SJ, Grossberg GT. Memantine: A review of studies into its safety and efficacy in treating Alzheimer’s disease and other dementias. Clin Interv Aging. 2009; 4: 367-377.
- Imbimbo BP, Giardina GA. y-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: Disappointments and hopes. Curr Top Med Chem. 2011; 11: 1555-1570.
- Sacena U. Alzheimer’s disease amyloid hypothesis at crossroads: Where do we go from here? Expert Opin Ther Targets. 2010; 14: 1273-1277.
- Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, et al. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or modetate Alzheimer’s disease. J Alzheimers Dis. 2015; 44: 705-720.
- Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid- beta-protein. J Alzheimers Dis. 2001; 3: 75-80.
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985; 82: 4245-4249.
- Selkoe DJ. Biochemistry and molecular biology of amyloid-beta-protein and the mechanism of Alzheimer’s disease. Handb Clin Neurol. 2008; 89: 245- 260.
- Cummings JL. Biomarkers in Alzheimer’s disease drug development. Alzheimers Dement. 2011; 7: e13-44.
- Herrmann N, Chau SA, Kircanski I, Lanct?t L. Current and emerging drug treatment options for Alzheimer’s disease. Drugs. 2011; 71: 2031-2056.
- Rosenmann H. CSF biomarkers for amyloid and tau pathology in Alzheimer’s disease. J Mol Neurosci. 2012; 47: 1-14.
- Brayne C, Matthews FE, Xuereb JH. Pathological correlates of late onset dementia in a multicenter, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet. 2001; 357: 169-175.
- Hanes DS, Weir MR. Usefulness of ARBs and ACE inhibitors in the prevention of vascular dementia in the elderly. Am J Geriatr Cardiol. 2007; 16: 175-182.
- Chen M, Maleski JJ, Sawmiller DR. Scientific truth or false hope: Understanding Alzheimer’s disease from an aging perspective. J Alzheimers Dis. 2011; 24: 3-10.
- Polidori MC, Pientka L. Bridging the pathophysiology of Alzheimer’s disease with vascular pathology: The feed-back, the feed-forward, and oxidative stress. J Alzheimers Dis. 2012; 28: 1-9.
- Hurd MD, Martorell P, Dalevande A, Mullen KJ, Panga M. Monetary costs of dementia in the United States. N Engl J Med. 2013; 368: 1326-1334.
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997; 18: 351-357.
- Hertze J, Minthon L, Zetterberg H, Vanmechelen E, Blennow K, Hansson O. Evaluation of CSF biomarkers as predictors of Alzheimer’s disease: A clinical follow-up study of 4.7 years. J Alzheimers Dis. 2010; 21: 1119-1128.
- Engelborghs S, Le Bastard N. The impact of cerebrospinal fluid biomarkers on the diagnosis of Alzheimer’s disease. Mol Diagn Ther. 2012; 16: 135-141.
- Anand S, Barnes JM, Young SA, Garcia DM, Tolley HD, Kauwe JSK, et al. Discovery and confirmation of diagnostic serum lipid biomarkers for Alzheimer’s disease using direct infusion mass spectrometry. J Alzheimers Dis. 2017; 59: 277-290.
- Bao W, Jia H, Fennema S, Cai Z, Carson RE, Huang YH. PET imaging for early detection of Alzheimer’s disease: From pathologic to physiologic biomarkers. PET Clin. 2017; 12: 329-350.
- Chen G, Shu H, Chen G, Ward BD, Antuono PG, Zhang Z, et al. Staging Alzheimer’s disease risk by sequencing brain function and structure, cerebrospinal fluid, and cognition biomarkers. J Alzheimers Dis. 2016; 54: 983-993.
- Schindler SE, Jasielec MS, Weng H, Hassenstab JJ, Grober E, McCue LM, et al. Neuropsychologiccal measures that detet early impairment and decline in preclinical Alzheimer disease. Neurobiol Aging. 2017; 56: 25-32.
- de la Torre JC. Carotid artery ultrasound and echocardiography testing to lower the prevalence of Alzheimer’s disease. J Stroke Cerebrovasc Dis. 2009; 18: 319-328.
- Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease. Neurology. 2009; 72: S1-S136.
- Ehringer H, Horaykieuicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin Wochenschr. 1960; 38: 1236-1239.
- Schapira AH, DmDermott MP, Barone P, Comella CL, Albrecht S, Hsu HH, et al. Pramipexole in patients with early Parkinson’s disease (PROUD): A randomized delayed-start trial. Lancet Neural. 2013; 12: 747-755.
- Welchko RM, Leveque XT, Dunbar GI. Genetic rat models of Parkinson’s disease. Parkinson’s Dis. 2012: 128356.
- Witte ME, Geurts JG, deVries HE, van der Valk, P, van Horssen J. Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion. 2010; 10: 411-418.
- Tufekci KU, Genc S, Genc K. The endotoxin-induced neuroinflammation model of Parkinson’s disease. Parkinson’s Dis. 2011; 2011: 487450.
- Nyholm D. Duodenal levodopa infusion monotherapy vs. oral polypharmacy in advanced Parkinson disease. Neurology. 2005; 64: 216-223.
- Deuschl G, Schade-Brittinger C, Krack P, Valkmann, J, Schafer H, Botzel K. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2006; 355: 896-908.
- Garcia Ruiz PJ. Efficacy of long-term continuous subcutaneous apomorphine infusion in advanced Parkinson’s disease with motor fluctuations: A multicenter study. Mov Disord. 2008; 23: 1130-1136.
- Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, et al. Priorities in Parkinson;s disease research. Nature Rev Drug Disc. 2011; 10: 377-393.
- Chaudhuri KR, Shapira AH. The non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009; 8: 464-474.
- Chaudhuri KR, Odin P, Antonini A, Martinez-Martin P. Parkinson’s disease: The non-motor issues. Parkinsonism Relat Disord. 2011; 17: 717-723.
- Dirnberger G, Jahanshahi M. Executive dysfunction in Parkinson’s disease; a review. J Neuropsychol. 2013; 7: 193-224.
- Lipski J, Nistico R, Berretta N, Guatteo E, Bernardi G, Mercuri NB. A scapegoat for accelerated neurodegeneration in Parkinson’s disease? Pro. Neurobiol. 2011; 94: 389-407.
- Allen AM, MacGregor DP, Chai SY, Donnan GA, Daczmarczyk S, Richardson K, et al. Angiotensin II receptor binding associated with nigrostriatal dopaminergic neurons in human basal ganglia. Ann Neurol. 1992; 32: 339-344.
- Re RN. Mechanisms of disease: Local renin-angiotensin-aldosterone systems and the pathogenesis and treatment of cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2004; 1: 42-47.
- Ruiz-Ortega M, Lorenzo O, Suzuki Y, Rupérez M, Egido J. Proinflammatory actions of angiotensins. Curr Opin Nephrol Hypertens. 2001; 10: 321-329.
- Suzuke Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochm Cell Biol. 2003; 35: 881-900.
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ Res. 2000; 86: 494-501.
- Munzel T, Kearney FJ. Are ACE inhibitors a “magic bullet” against oxidative stress? Circulation. 2001; 104: 1571-1574.
- Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: Upstream mediators. Circ Res. 2002; 91: 406-413.
- Babior BM. NADPH oxidase. Curr Opin Immunol. 2004; 16: 42-47.
- Joglar B, Rodriguez-Pallares J, Rodriguez-Peres AI, Rey P, Guerra MJ, Labandeira-Garcia JL. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J Neurochem. 2009; 109: 656-669.
- Okamura A, Rakugi H, Ohishi M, Yanagitani Y, Takiuchi S, Moriguchi K, et al. Upregulation of renin-angiotensin system during differentiation of monocytes to macrophages. J Hypertens. 1999; 17: 537-545.
- Lenkei Z, Palkovits M, Corvol P, Llorens-Cortes C. Expression of angiotensin type-1 (AT1) and type-2 (AT2) receptor mRNAs in the adult rat brain: A functional neuroanatomical review. Front Neuroendocrinol. 1997; 18: 383- 439.
- Simonnet G, Giorguieff-Chesselet MF, Carayon A, Bioulac B, Cesselin F, Glowinski J, et al. Angiotensin II and nigrostriatal system. J Physiol (Paris). 1981; 77: 71-79.
- Chai SY, Christie MJ, Beart PM, Mendelsohn FA. Effects of Nigral dopaminergic lesions and striatal excitotoxin lesions on brain converting enzyme. Neurochem Int. 1987; 10: 101-107.
- Chai SY, McKenzie JS, McKinley MJ, Mendelsohn FA. Angiotensin converting enzyme in the human basal forebrain and midbrain visualized by in vitro autoradiography. J Comp Neurol. 1990; 291: 179-194.
- Strittmatter SM, Thiele EA, Kapiloff MS, Snyder SH. A rat brain isozyme of angiotensin-converting enzyme. Unique specificity for amidated peptide substrates. J Biol Chem. 1985; 260: 9825-9832.
- Reardon KA, Mendelsohn FA, Chai SY, Horne MK. The Angiotensin Converting Enzyme (ACE) inhibitor, perindopril, modifies the clinical features of Parkinson’s disease. Aust N Z J Med. 2000; 30: 48-53.
- Jenkins TA, Wong JY, Howells DW, Mendelsohn A, Chai SY. Effect of chronic angiotensin-converting enzyme inhibition on striatal dopamine content in the MPTP-treated mouse. J Neurochem. 1999; 73: 214-219.
- Ehlers MR, Riordan JF. Angiotensin-converting enzyme: New concepts concerning its biological role. Biochemistry. 1989; 28: 5311-5318.
- Rodriguez-Pallares J, Rey P, Parga JA, Munoz A, Guerra MJ, Labandeira Garcia JL. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol Dis. 2008; 31: 58-73.
- Munoz A, Rey P, Guerra MJ. Reduction of dopaminergic degeneration and oxidative stress by inhibition of angiotensin converting enzyme in a MPTP model of Parkinsonism. Neuropharmacology. 2006; 51: 112-120.
- Lopez-Real A, Rey P, Soto-Otero R, Mendez-Alvarez E, Labandeira-Garcia JL. Angiotensin-converting enzyme inhibition reduces oxidative stress and protects dopaminergic neurons in a 6-hydroxydopamine rat model of Parkinsonism. J Neurosci Res. 2005; 81: 865-873.
- Mertens B, Vanderheyden P, Michott Y, Sarre S. The role of the central renin-angiotensin system in Parkinson’s disease. J Renin Angiotensin Aldosterone Syst. 2010; 11: 49-56.
- Labandeira-Garcia JL, Rodriquez-Pallares J, Villar-Cheda B, Rodriquez-Perz AI, Garrido-Gil P, Guerra MJ. Aging, angiotensin system and dopaminergic degeneration in the substantia nigra. Aging Dis. 2011; 2: 257-274.
- Rey P, Lopez-Real A, Sanchez-Iglesias S, Munoz A, Soto-Otero R, Labandeira-Garcia JL. Angiotensin type-1-receptor antagonists reduce 6-hydroxy-dopamine toxicity for dopaminergic neurons. Neurobiol Aging. 2007; 28: 555-567.
- Grammatopoulos TN, Jones SM, Ahmadi FA, Hoover BR, Snell LD, Skoch J, et al. Angiotensin type 1 receptor antagonist losartan, reduces MPTPinduced degeneration of dopaminergic neurons in substantia nigra. Mol Neurodegen. 2007; 2: 1-17.
- Becker C, Jick SS, Meier CR. Use of antihypertensives and the risk of Parkinson Disease. Neurology. 2008; 70: 438-444.
- Ascherio A, LeWitt PA, Xu K, Eberly S, Watts A, Matson WR. Urate predicts rate of clinical decline in Parkinson disease. Arch Neurol. 2009; 66: 1460- 1468.
- Grammatopoulos TN, Outeiro TF, Hyman BT, Standaert DG. Angiotensin II protects against a-synuclein toxicity and reduces protein aggregation in vitro. Biochem Biophy Res Comm. 2007; 363: 846-851.
- Collier TJ, Kipton J, Daley F, Palfi S, Chu Y, Sortwell C, et al. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: Diminished compensatory mechanisms as a prelude to Parkinsonism. Neurobiol Dis. 2007; 26: 56-65.
- Cruz-Muros I, Afonso-Oramas D, Abreu P, Perez-Delgado MM, Rodriguez M, Gonsalez-Hernandez T. Aging effects on the dopamine transporter expression and compensatory mechanisms. Neurobiol Aging. 2009; 30: 973-986.
- Villar-Cheda B, Valenzuela R, Rodriguez-Perez AI, Guerra MJ, Labandeira- Garcia JL. Aging-related changes in the nigral angiotensin system enhances proinflammatory and pro-oxidative markers and 6-OHDA-induced dopaminergic degeneration. Neurobiol Aging. 2012; 33: 204.e1-11.
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002; 22: 1763-1771.
- Rodriguez-Perez AI, Valenzuela R, Joglar B, Garrido-Gil P, Guerra MJ, Labandeira-Garcia JL. Renin angiotensin system and gender differences in dopaminergic degeneration. Mol Neurodegener. 2011; 6: 58.
- Zawada WM, Banninger BP, Thornton J, Marriott B, Cantu D, Rachubinski AL, et al. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidasedependent two-wave cascade. J. Neuroinflamm. 2011; 8: 129.e.1-13.
- Touyz RM, Chen X, Tabet F, Yao GH, Quinn T, Pagano PJ, et al. Expression of a functionally active gp91 phox-containing neurtrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res. 2002; 90, 1205-1213.
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004; 279: 1415-1421.
- Stragier B, De Bundel D, Sarre S, Smolders I, Vauguelin G, Dupont A, et al. Involvement of insulin-regulated aminopeptidase in the effects of the reninangiotensin fragment angiotensin IV: A review. Heart Fail Rev. 2008; 13: 321-337.
- Stragier B, Sarre S, Vanderheyden P, Vauquelin G, Fournie-Zaluski MC, Ebinger G, et al. Metabolism of angiotensin II is required for its in vivo effect on dopamine release in the striatum of the rat. J Neurochem. 2004; 90: 1251-1257.
- Nakamura T, Muzuno S, Matsumoto K, Sawa Y, Matsuda H, Nakamura T. Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J Clin Invest. 2000; 106: 1511-1519.
- Van Belle E, Witzenbichler B, Chen D, Silver M, Chang L, Schwall R, et al. Potentiated angiogenic effect of scatter factor/hepatocyte growth factor via induction of vascular endothelial growth factor: The case for paracrine amplification of angiogenesis. Circulation. 1998; 97: 381-390.
- Morishita R, Nakamura S, Hayashi S, Taniyama Y, Moriguchi, Nagano, T, et al. Therapeutic angiogenesis induced by human recombinant hepatocyte growth factor in rabbit hind limb ischemia model as cytokine supplement therapy. Hypertension. 1999; 33: 1379-1384.
- Miyazawa T, Matsumoto K, Ohmichi H, Katoh H, Yamashima T, Hakamura T. Protection of hippocampal neurons from ischemia-induced delayed neuronal death by hepatocyte growth factor: A novel neurotrophic factor. J Cereb Blood Flow Metab. 1998; 18: 345-348.
- Tsuzuki N, Miyazawa T, Matsumoto K, Nakamura T, Shima K. Hepatocyte growth factor reduces the infarct volume after transient focal cerebral ischemia in rats. Neurol. Res. 2001; 23, 417-424.
- Date I, Takagi N, Takagi K, Kago T, Matsumoto K, Nakamura T, et al. Hepatocyte growth factor improved learning and memory dysfunction of microsphere-embolized rats. J Neurosci Res. 2004; 78: 442-543.
- Shimamura M, Sato N, Waguri S, Uchiyama Y, Hayashi T, Iida H, et al. Gene transfer of hepatocyte growth factor gene improves learning and memory in the chronic stage of cerebral infarction. Hypertension. 2006; 47: 742-751.
- Kramár EA, Krishnan R, Harding JW, Wright JW. Role of nitric oxide in angiotensin IV-induced increases in cerebral blood flow. Regul Pept. 1998; 74: 185-192.
- Papazafiropoulou AK, Koros C, Melidonis A. Antonopoulos S, Diabetes and dementia – the two faces of Janus. Arch Med Sci – Anterosc Dis. 2020; 2020: 5:e186-e197.
- Verdile G, Fuller SJ, Martins RN. The role of type 2 diabetes in neurodegeneration. Neurobiol Dis. 2015; 84: 22-38.
- Simo R, Ciudin A, Simo-Servat O, Hernandez C. Cognitive impairment and dementia: A new emerging complication of type 2 diabetes – The diabetologist’s perspective. Acta Diabeetol. 2017; 54: 417-424.
- Bello-Chavolla OY, Antonio-Villa NE, Vargas-Vazquez A, Avila-Funes JA, Aguilar-Salinas CA. Pathophysiological mechanisms linking type 2 diabetes and dementia: Review of evidence from clinical, translational and epidemiological research. Curr Diabetes Rev. 2019; 15: 456-470.
- Koekkoek P, Kappelle LJ, van den Berg E, Rutten GE, Biessels GJ. Cognitive function in patients with diabetes mellitus: Guidance for daily care. Lancet Neurol. 2015; 14: 329-340.
- Tramutola A, Lanzillotta C, Perluigi M, Butterfield DA. Oxidative stress, protein modification and Alzheimer disease. Brain Res Bull. 2017; 133: 88- 96.
- Irwin K, Sexton C, Daniel T, Lawlor B, Naci L. Healthy aging and dementia: Two roads diverging in midlife? Front Aging Neurosci. 2018; 20: 275-295.
- Wright JW, Harding JW. Contributions by the brain renin-angiotensin system to memory, cognition, and Alzheimer’s disease. J Alzheimers Dis. 2019; 67: 469-480.
- Chen JJ, Wang T, An CD, Jiang CY, Zhao J, Li S. Brain derived neurotrophic factor: A mediator of inflammation-associated neurogenesis in Alzheimer’s disease. Rev Neurosci. 2016; 27: 793-811.
- Fukumoto M, Takai S, Ishizaki E, Sugivama T, Oku H, Jin D, et al. Involvement of angiotensin II-dependent vascular endothelial growth factor gene expression via NADPH oxidase in the retina in a type 2 diabetic rat model. Cur Eye Res. 2008; 33: 885-891.
- Steckelings UM, Rompe F, Kaschina E, Unger T. The evolving story of the RAAS in hypertension, diabetes and CV disease – moving from macrovascular to microvascular targets. Fundam Clin Pharmacol. 2009; 23: 693-703.
- Luther JM, Brown NJ. The renin-angiotensin-aldosterone system and glucose homeostasis. Trends Pharmacol Sci. 2011; 32: 734-739.
- Funatsu H, Yamashita H, Ikeda T, Nakanishi Y, Kitano S, Hori S. Angiotensin II and vascular endothelial growth factor in the vitreous fluid of patients with diabetic macular edema and other retinal disorders. Amer J Ophthalmol. 2002; 133: 537-543.
- Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: Effects of ACEI and ARB. Kidney Internat. 2002; 61: 186-194.
- Mauer M, Zinman B, Gardiner R, Drummond KN, Suissa S, Donnelly SM, et al. ACE-I and ARBs in early diabetic nephropathy. J Renin Angiotensin Aldosterone Syst. 2002; 3: 262-269.
- Chiarelli F, Di Marzio D, Santilli DF, Mohn A, Blasetti A, Cipollone F, et al. Effects of irbesartan on intracellular antioxidant enzyme expression and activity in adolescents and young adults with early diabetic angiopathy.” Diabetes Care. 2005; 7: 1690-1697.
- Izuhara Y, Sada T, Yanagisawa H, Koike H, Ohtomo S, Dan T, et al. Novel sartan derivative with very low angiotensin II type 1 receptor affinity protects the kidney in type 2 diabetic rats. Arterioscl Thrombotic Vasc Biol. 2008; 28: 1767-1773.
- Chaturvedi N, Porta M, Klein R, Orchard T, Fuller J, Parving HH, et al. DIRECT Programme Study Group, “Effect of candesartan on prevention (DIRECT-Prevent 1) and progression (DIRECT-Protect 1) of retinopathy in type 1 diabetes: Randomized, placebo-controlled trials,” Lancet. 2008; 372: 1394-1402.
- Bichu P, Nistala R, Khan A, Sowers JR, Whaley-Connell A. Angiotensin receptor blockers for the reduction of proteinuria in diabetic patients with overt nephropathy: Results from the AMADEO study.” Vasc Health Risk Manag. 2009; 5: 129-140.
- Chen P, Guo AM, Edwards PA, Trick G, Scicli AG. Role of NADPH oxidase and ANG II in diabetes-induced retinal leukostasis. Amer J Physiol, Reg Integr Comp Physiol. 2007; 293: R1619-1629.
- Sjølie AK, Klein R, Porta P, Orchard T, Fuller J, Parving HH, et al. DIRECT Programme Study Group, Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (DIRECT-Protect2): A randomized placebo-controlled trial. Lancet. 2008; 372: 1385-1393.
- Dzau V, Braunwald E. Resolved and unresolved issues in the prevention and treatment of coronary artery disease: A workshop consensus statement. Am Heart J. 1991; 121: 1244-1263.
- Gebre AK, Altaye BM, Atey TM, Tuem KB, Berhe DF. Targeting reninangiotensin system against Alzheimer’s disease. Front Pharmacol. 2018; 9: 440-454.
- Morley JE. Diabetes and aging: Epidemiologic overview. Clin Geriatr Med. 2008; 24: 395-405.
- Topper R, Gehrmann J, Banati R, Schwartz M, Block F, Noth J. Rapid appearance of beta-amyloid precursor protein immunoreactivity in glial cells following excitotoxic brain injury. Acta Neuropathol. 1995; 89: 23-28.
- Chatterjee D, Kordower JH. Immunotherapy in Parkinson’s disease: Current status and future directions. Neurobiol Dis. 2019; 12: 104587.
- Petrie JR, Guzik TJ, Touyz RM. Diabetes, hypertension, and cardiovascular disease: Clinical insights and vascular mechanisms. Canad J Cardiol. 2018; 34: 575-584.
- Hoessli C, Johnson JD, Piret JM. Purified human pancreatic duct cell culture conditions defined by serum-free high content growth factor screening. PloS One. 2012; 7: e33999.
- Oliveira AG, Araujo TG, de Melo Carvalho B, Rocha GZ, Santos A, Saad MJ. The role of Hepatocyte Growth Factor (HGF) in insulin resistance and diabetes. Front Endocrinol (Lausanne). 2018; 9: 503.
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR. “The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R),” JAMA. 2003; 289: 3095-3105.
- Kessler RC, Avenevoli S, Merikangas K. Mood disorders in children and adolescents: An epidemiologic perspective. Biol Psychiatry. 2001; 49: 1002- 1014.
- Anxiety and Depression Association of America. National Institute of Mental Health. 2020.
- Dozeman E, van Marwijk HW, van Schaik DJ, Stek ML, van der Horst HE, Beekman AT. High incidence of clinically relevant depressive symptoms in vulnerable persons of 75 years or older living in the community. Aging Ment Health. 2010; 14: 828-833.
- Meeks TW, Vahia IV, Lavretsky H, Jesle DV. A tune in “a minor” can “b major”: A review of epidemiology, illness course, and public health implications of subthreshold depression in older adults. J Affect Disord. 2010; 129: 126-42.
- Thielke SM, Diehr P, Unutzer J. Prevalence, incidence, and persistence of major depressive symptoms in the Cardiovascular Health Study. Aging and Mental Health 2010; 14: 168-176.
- Fang J, Cheng Q. Etiological mechanisms of post-stroke depression: A review. Neurol Res. 2009; 31: 904-909.
- Rodriguez-Oroz MC, Jahanshahi M, Krack P, Litvan I, Macias R, Bezard E. Initial clinical manifestations of Parkinson’s disease: Features and pathophysiological mechanisms. Lancet Neurol. 2009; 8: 1128-1139.
- Aznar S, Knudsen GM. Depression and Alzheimer’s disease: Is stress the initiating factor in a common neuropathological cascade? J Alzheimers Dis. 2011; 23: 177-193.
- Bremmer MA, Beekman AT, Deeg DJ, Penninx BW, Dik MG, Hack CE. Inflammatory markers in late-life depression: Results from a populationbased study. J Affect Disord. 2008; 106: 249-255.
- Flicker L. Cardiovascular risk factors, cerebrovascular disease burden, and healthy brain aging. Clin Geriatr Med. 2010; 26: 17-27.
- Zubenko GS, Nixon RA. Mood elevating effects of captopril in depressed patients. Am J Psychiatry. 1984; 141: 110-111.
- Deicken RF. Captopril treatment of depression. Biol Psychiatry. 1986; 12: 1425-1428.
- Germain L, Chouinard G. Treatment of recurrent unipolar major depression with captopril. Iol Psychiatry. 1988; 23: 637-641.
- Germain L, Chouinard G. Captopril treatment of major depression with serial measurements of blood cortisol concentrations. Bio Psychiatry. 1989; 25: 489-493.
- Goldstein JM, Knobloch-Litwin LC, Malick JB. Behavioural evidence for β-adrenoceptor subsensitivity after subacute antidepressant/a2- adrenoceptor antagonist treatment. Naunyn-Schmiedeberg’s Arch Pharmacol. 1985; 329: 355-358.
- Przegalinski E, Siwanowicz J, Baran L. Effect of repeated administration of antidepressant drugs on the isoprenaline-induced drinking in rats. Pol J Pharmacol. 1988; 40: 251-258.
- Gard PR, Mycroft N. Reduction of angiotensin II-induced drinking in rats by 21-hour pretreatment with desipramine. J Pharm Pharmacol. 1991; 43: 690- 693.
- Gard PR, Mandy A, Whiting JM, Nickels DP, Meakin AJ. Reduction of responses to angiotensin II by antidepressant drugs. Eur J Pharmacol. 1994; 264: 295-300.
- Giardina WJ, Ebert DM. Positive effects of captopril in the behavioural despair swim test. Biol Psychiatry. 1989; 25: 697-702.
- Martin P, Massol J, Puech AJ. Captopril as an antidepressant? Effects on the learned helplessness paradigm in rats. Biol Psychiatry. 1990; 27: 968- 974.
- Gard PR, Mandy A, Surcliffe MA. Evidence of a possible role of altered angiotensin function in the treatment, but not aetiology, of depression. Biol Psychiatry. 1999; 45: 1030-1034.
- Gard PR. The role of angiotensin II in cognition and behavior. Eur J Pharmacol. 2002; 438: 1-14.
- Park HS, You MJ, Yang B, Jang KB, Yoo J, Choi HJ, et al. Chronically infused angiotensin II induces depressive like behavior via microglia activation. Nature Res. 2020; 10: 22082.
- Arnold SE, Xie SX, Leung YY, Wang LS, Kling MA, Han X, et al. Plasma biomarkers of depressive symptoms in older adults. Transl Psychiatry. 2012; 2: ID e65,10.1038/tp.2011.63.
- Hucimusalar Y, Esel E. Suggested biomarkers for major depressive disorder. Noro Psikiyatr Ars. 2018; 55: 280-290.
- Kalkman HO. The association between vascular inflammation and depressive disorder. Causality, biomarkers and targeted treatment. Pharmaceuticals. (Basel) 2020; 13: 92.
- Bremmer MA, Deeg DJ, Beekman AT, Pelnnix BW, Lips P, Hoogendijk WJ. Major depression in late life is associated with both hypo-and hypercortisolemia. Biol Psychiatry. 2007; 62: 479-486.
- Amato L, Paolisso G, Cacciatore F, Ferrara N, Canonico S, Grengo F. Noninsulin- dependent diabetes mellitus is associated with a greater prevalence of depression in the elderly. The Osservatorio Geriatrico of Campania Region Group. Diabetes Metab. 1996; 22: 314-318.
- Lee KS, Chung JH, Lee KH, Shin MJ, Oh BH, Lee SH, et al. Simultaneous measurement of 23 plasma cytokines in late-life depression. Neurol Sci. 2009; 30: v435-438.
- Feltes PK, Doorduin J, Klein HC, Juarez-Orozco LE, Dierckx RA, Moriguchi- Jeckel CM, et al. Anti-inflammatory treatment for major depressive disorder: Implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J Psychopharmacol. 2017; 31: 1149- 1165.
- Diniz BS, Teixeira AL, Talib LL, Mendonca VA, Gattaz WF, Forlenza OV. Serum brain-derived neurotrophic factor level is reduced in antidepressantfree patients with late-life depression. World J Biol Psychiatry. 2010; 11: 550-555.
- Ho N, Sommers MS, Lucki I. Effects of diabetes on hippocampal neurogenesis: Links to cognition and depression. Neurosci Biobehav Rev. 2013; 37: 1346-1362.
- Stuart MJ, Baune BT. Depression and type 2 diabetes: Inflammatory mechanisms of a psychoneuroendocrine co-morbidity. Neurosci Biobehav Rev. 2012; 36: 658-676.
- Katon W, Pedersen HS, Ribe AR. Effect of depression and diabetes mellitus on the risk for Dementia: A national population-based cohort study. JAMA Psychiatry. 2015; 72: 612-619.
- Mill J, Petronis A. Molecular studies of major depressive disorder: The epigenetic perspective. Mol Psychiatry. 2007; 12: 799-814.
- Miguel J, Hidalgo H, Rajkowska G. Morphological brain changes in depression: Can antidepressants reverse them. CNS Drugs. 2002; 16: 361- 372.
- Drevets W, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct Funct. 2008; 213: 93-118.
- Belleau EL, Treadway MT, Pizzagalli DA. The impact of stress and major depressive disorder on hippocampal and medial prefrontal cortex morphology. Biol Psychiatry. 2019; 85: 443-453, 2019.
- Singh MK. Anomalous gray matter structural networks in major depressive disorder. Biol Psych. 2013; 74: 777-785.
- Luo Q, Deng Z, QinJ, Wei D, Cun L, Qiu J, et al. Frequency dependent topological alterations of intrinsic functional connectome in major depressive disorder. Sci Rep. 2015; 5: 9710.
- Jiang X, Shen Y, Yao J, Zhang L, Xu L, Geng R. Connectome analysis of functional and structural hemispheric brain networks in major depressive disorder. Transl Psychiatry. 2019; 9: 136.
- Gong Q, He Y. Depression, neuroimaging and connectomics: A selective overview. Biol Psych. 2015; 77: 223-235.
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006; 59: 1116-1127.
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature 2008; 455: 894-902.
- Amrein I, Isler K, Lipp HP. Comparing adult hippocampal neurogenesis in mammalian species and orders: Influence of chronological age and life history stage. Eur J Neurosci. 2011; 34: 978-987.
- Duman RS, Li N. A neurotrophic hypothesis of depression: Role of synaptogenesis in the actions of NMDA receptor antagonists. Philos Trans R Soc Lond B Biol Sci. 2012; 367: 2475-2484.
- Drapeau E, Abrous DN. Stem cell review series: Role of neurogenesis in age-related memory disorders. Aging Cell. 2008; 7: 569-589.
- Miranda CJ, Braun L, Jiang Y, Hesler ME, Zhang L, Riolo M, et al. Aging brain microenvironment decreases hippocampal neurogenesis through Wntmediated surviving signaling. Aging Cell. 2012; 11: 542-552.
- Boldrini M, Underwood MD, Hen R, Rosoklia OB, Dwork AJ, John Mann J, et al. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009; 34: 2376-2389.
- Serafini G, Hayley S, Pompili M, Dwivedi Y, Brahmachari G, Girardi P, et al. Hippocampal neurogenesis, neurotrophic factors and depression: Possible therapeutic targets? CNS & Neurological Dis – Drug Targets. 2014; 13: 1708-1721.
- Fournier NM, Duman RS. Role of vascular endothelial growth factor in adult hippocampal neurogenesis: Implications for the pathophysiology and treatment of depression. Behav Brain Res. 2011; 227: 440-449.
- Masi G, Brovedani P. The hippocampus, neurotrophic factors and depression: Possible implications for the pharmacotherapy of depression, CNS Drugs. 2011; 25: 913-931.
- Hayley S, Litteljohn D. Neuroplasticity and the next wave of antidepressant strategies. Front Cell Neurosci. 2013; 7: 1-17.
- Calabrese F, Rossetti AD, Racagni G, Gass P, Riva MA, Molteni R. Brain-derived neurotrophic factor: A bridge between inflammation and neuroplasticity. Front Cell Neurosci. 2014; 8: 1-7.
- Sharma AN, Borges da Costa e Silva BF, Soares JC, Carvalho AF, Quevedo J. Role of trophic factors GDNF, IGF-1 and VEGF in major depressive disorder: A comprehensive review of human studies. J Affect Disord. 2016; 197: 9-20.
- Castren E, Rantamaki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev Neurobiol. 2010; 70: 2892-2897.
- Heldt S, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007; 12: 656-670.
- Taliz D, Stall N, Dar DE, Zangen A. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry. 2010; 15: 80-92.
- Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. BDNF Val66met allele impairs basal and Ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012; 71: 996-1005.
- AMD: The Latest Results from the AREDS2 Study. 2021.
- Diabetes caucus-degette. House. 2021.
- National Advisory Eye Council Meeting. 2021.
- Giese MJ, Speth RC. The ocular renin-angiotensin system: A therapeutic target for the treatment of ocular disease. Pharmacol Ther. 2014; 142: 11- 32.
- Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006; 86: 747-803.
- Holappa M, Vapaatalo H, Vaajanen. Local ocular renin-angiotensinaldosterone system: Any connection with intraocular pressure? A comprehensive review. Ann Med. 2020; 42: 191-206.
- Janssen SF, Gorgels TG, van der Spek PJ. In silico analysis of the molecular machinery underlying aqueous humor production: Potential implications for glaucoma. J Clin Bioinformatics. 2013; 3: 21.
- Buys ES, Potter LR, Pasquale LR. Regulation of intraocular pressure by soluble and membrane guanylate cyclases and their role in glaucoma. Front Mol Neurosci. 2014; 7: 38.
- Vieira GM, Oliveira HB, de Andrade DT. Intraocular pressure variation during weight lifting. Arch Ophthalmol. 2006; 124: 1251-1254.
- Goel M, Picciani RG, Lee RK. Aqueous humor dynamics: A review. TOOPHTJ 2010; 4: 52-59.
- Levin LA, Crowe ME, Quigley HA. Neuroprotection for glaucoma: Requirements for clinical translation. Exp Eye Res. 2017; 157: 34-37.
- Tian B, Geiger B, Epstein DL. Cytoskeletal involvement in the regulation of aqueous humor outflow. Invest Ophthalmol Vis Sci. 2000; 41: 619-623.
- Tian B, Gabelt BT, Geiger B. The role of the actomyosin system in regulating trabecular fluid outflow. Exp Eye Res. 2009; 88: 713-717.
- Tan JC, Peters DM, Kaufman PL. Recent developments in understanding the pathophysiology of elevated intraocular pressure. Curr Opin Ophthalmol. 2006; 17: 168-174.
- Vranka JA, Kelley MJ, Acott TS. Extracellular matrix in the trabecular meshwork: Intraocular pressure regulation and dysregulation in glaucoma. Exp Eye Res. 2015; 133: 112-125.
- Vranka JA, Acott TS. Pressure-induced expression changes in segmental flow regions of the human trabecular meshwork. Exp Eye Res. 2017; 158: 67-72.
- Andrés-Guerrero V, García-Feijoo J, Konstas AG. Targeting schlemm’s canal in the medical therapy of glaucoma: Current and future considerations. Adv Ther. 2017; 34: 1049-1069.
- Brubaker RF. The flow of aqueous humor in the human eye. Trans Am Ophthalmol Soc. 1982; 80: 391-474.
- Civan MM, Macknight AD. The ins and outs of aqueous humour secretion. Exp Eye Res. 2004; 78: 625-631.
- Marcus DF, Krupin T, Podos SM. The effect of exercise on intraocular pressure. I. human beings. Invest Ophthalmol. 1970; 9: 749-752.
- Dane S, Koçer I, Demirel H. Long-term effects of mild exercise on intraocular pressure in athletes and sedentary subjects. Int J Neurosci. 2006; 116: 1207-1214.
- Dane S, Koçer I, Demirel H. Effect of acute submaximal exercise on intraocular pressure in athletes and sedentary subjects. Int J Neurosci. 2006; 116: 1223-1230.
- Baskaran M, Raman K, Ramani KK. Intraocular pressure changes and ocular biometry during sirsasana (headstand posture) in yoga practitioners. Ophthalmology. 2006; 113: 1327-1332.
- Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004; 363: 1711-1720.
- King A, Tuulonen A, Azuara-Blanco A. Glaucoma. Brit Med J. 2013:1-9.
- Schmidl D, Schmetterer L, Garhöfer G. Pharmacotherapy of glaucoma. J Ocul Pharmacol Ther. 2015; 31: 63-77.
- Collaborative Normal-Tension Glaucoma Study Group: Comparison of glaucomatous progression between untreated patients with normal-tension glaucoma and patients with therapeutically reduced intraocular pressures. Am J Ophthalmol. 1998; 1264: 487-497.
- Collaborative Normal-Tension Glaucoma Study Group: The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Am J Ophthalmol. 1998; 1264: 498-505.
- Hirooka K, Baba T, Fujimura T. Prevention of visual field defect progression with angiotensin-converting enzyme inhibitor in eyes with normal-tension glaucoma. Am J Ophthalmol. 2006; 142: 523-525.
- Boland MV, Ervin AM, Friedman DS. Comparative effectiveness of treatments for open-angle glaucoma: A systematic review for the U.S. preventive services task force. Ann Intern Med. 2013; 158: 271-279.
- Conlon R, Saheb H, Ahmed I. Glaucoma treatment trends: A review. Can J Ophthalmol. 2017; 52: 114-124.
- Igic R, Robinson CJ, Milosevic Z, Wilson CM, Erdos EG. Activity of renin and angiotensin I converting enzyme in retina and ciliary body. Lijec Vjesn. 1977; 99: 482-484.
- Holappa M, Valjakka J, Vaajanen A. Angiotensin (1-7) and ACE2, “The Hot Spots” of renin-angiotensin system, detected in the human aqueous humor. TOOPHTJ. 2015; 9: 28-32.
- Holappa M, Vapaatalo H, Vaajanen A. Many faces of renin-angiotensin system - focus on eye. TOOPHTJ. 2017; 11: 122-142.
- Vaajanen A, Kalesnykas G, Vapaatalo H. The expression of Mas-receptor of the renin-angiotensin system in the human eye. Graefes Arch Clin Exp Ophthalmol. 2015; 253: 1053-1059.
- Igic R. Four decades of ocular renin-angiotensin and kallikrein-kinin systems (1977–2017). Exp Eye Res. 2018; 166: 74-83.
- Cullinane AB, Leung PS, Ortego J. Renin-angiotensin system expression and secretory function in cultured human ciliary body non-pigmented epithelium. Br J Ophthalmol. 2002; 86: 676-683.
- Vaajanen A, Vapaatalo H. Local ocular renin-angiotensin system - a target for glaucoma therapy? Basic Clin Pharmacol Toxicol. 2011; 109: 217-224.
- Watkins RW, Baum T, Cedeno K. Topical ocular hypotensive effects of the novel angiotensin converting enzyme inhibitor SCH 33861 in conscious rabbits. J Ocul Pharmacol. 1987; 3: 295-307.
- Costagliola C, Di Benedetto R, De Caprio L. Effect of oral captopril (SQ 14225) on intraocular pressure in man. Eur J Ophthalmol. 1995; 5: 19-25.
- Costagliola C, Verolino M, De Rosa ML. Effect of oral losartan potassium administration on intraocular pressure in normotensive and glaucomatous human subjects. Exp Eye Res. 2000; 71: 167-171.
- Shah GB, Sharma S, Mehta AA. Oculohypotensive effect of angiotensinconverting enzyme inhibitors in acute and chronic models of glaucoma. J Cardiovasc Pharmacol. 2000; 36: 169-175.
- Inoue T, Yokoyoma T, Mori Y. The effect of topical CS-088, an angiotensin AT1 receptor antagonist, on intraocular pressure and aqueous humor dynamics in rabbits. Curr Eye Res. 2001; 23: 133-138.
- Wang RF, Podos SM, Mittag TW. Effect of CS-088, an angiotensin AT1 receptor antagonist, on intraocular pressure in glaucomatous monkey eyes. Exp Eye Res. 2005; 80: 629-632.
- Mehta A, Iyer L, Parmar S. Oculohypotensive effect of perindopril in acute and chronic models of glaucoma in rabbits. Can J Physiol Pharmacol. 2010; 88: 595-600.
- Vaajanen A, Vapaatalo H, Kautiainen H. Angiotensin (1-7) reduces intraocular pressure in the normotensive rabbit eye. Invest Ophthalmol Vis Sci. 2008; 49: 2557-2562.
- Dias J, Axelband F, Lara LS. Is angiotensin-(3-4) (Val-Tyr), the shortest angiotensin II-derived peptide, opening new vistas on the renin-angiotensin system? J Renin Angiotensin Aldosterone Syst. 2017; 18: 147032031668933.
- Fitzsimons JT. Angiotensin stimulation of the central nervous system. Rev Physiol Biochem Pharmacol. 1980; 87: 117-167.
- Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev. 1998; 78: 583-686.
- Tonnaer JA, Wiegant VM, DeJong W, DeWied D. Central effects of angiotensin on drinking and blood pressure: Structure-activity relationships. Brain Res. 1982; 236: 417-428.
- Al-Zamil WM, Yassin SA. Recent developments in age-related macular degeneration: A review. Clin Intervent Aging. 2017; 12: 1313-1330.
- Parekh N, Voland RP, Moeller SM. CAREDS Research Study Group. Association between dietary fat intake and age-related macular degeneration in the Carotenoids in Age-Related Eye Disease Study (CAREDS): An ancillary study of the women’s health initiative. Arch Ophthalmol. 2009; 127: 1483-1493.
- Adams MK, Simpson JA, Aung KZ. Abdominal obesity and age-related macular degeneration. Am J Epidemiol. 2011; 173: 1246-1255.
- Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K. Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci. 2016; 73: 1765-1786.
- Ruan Y, Jiang S, Musayeva A, Gericke A. Oxidative stress and vascular dysfunction in the retina: Therapeutic strategies. Antioxidants. 2020; 9: 761.
- Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, et al. Retinopathy in diabetes. Diabetes Care. 2004; 27: S84-S87.
- Wilkinson CP, Ferris FL, Klein RE, Lee PP, Agardh CD, Davis M, et al. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology. 2003; 110: 1677-1682.
- Wakisaka M, Yoshinari M, Nakamura S, Asano T, Sonoki K, Shi Ah, et al. Suppression of sodium-dependent glucose uptake by captopril improves high-glucose-induced morphological and functional changes of cultured bovine retinal pericytes. Microvasc Res. 1999; 58: 215-223.
- Downie LE, Pianta MJ, Vingrys AJ, Wilkinson-Berka JL, Fletcher EL. AT1 receptor inhibition prevents astrocyte degeneration and restores vascular growth in oxygen-induced retinopathy. Glia. 2008; 56: 1076-1090.
- Verma A, Shan Z, Lei B, Yuan L, Liu X, Nakagawa T, et al. ACE2 and Ang- (1-7) confer protection against development of diabetic retinopathy. Mol Ther. 2012; 20: 28-36.
- Otani A, Takagi H, Suzuma K, Honda Y. Angiotensin II potentiates vascular endothelial growth factor-induced angiogenic activity in retinal microcapillary endothelial cells. Circ Res. 1998; 82: 619-628.
- Yamagishi S, Imaizumi T. Pericyte biology and diseases. Int J Tissue React. 2005; 27: 125-135.
- Wolff SP, Dean RT. Glucose autoxidation and protein modification. The potential role of ‘autoxidative glycosylation’ in diabetes. Biochem J. 1987; 245: 243-250.
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994; 74: 1141-1148.
- Mori F, Hikichi T, Nagaoka T, Takahashi J, Kitaya N, Yoshida A. Inhibitory effect of losartan, an AT1 angiotensin II receptor antagonist, on increased leucocyte entrapment in retinal microcirculation of diabetic rats. Br J Ophthalmol. 2002; 86: 1172-1174.
- Chen P, Scicli GM, Guo M, Fenstermacher JD, Dahl D, Edwards PA. Role of angiotensin II in retinal leukostasis in the diabetic rat. Exp Eye Res. 2006; 83: 1041-1051.
- Wilkinson-Berka JL, Tan G, Jaworski K, Ninkovic S. Valsartan but not atenolol improves vascular pathology in diabetic Ren-2 rat retina. Am J Hypertens. 2007; 20: 423-430.
- Zhang JZ, Xi X, Gao L, Kern TS. Captopril inhibits capillary degeneration in the early stages of diabetic retinopathy. Curr Eye Res. 2007; 32: 883-889.
- Sola A, Saldeño YP, Favareto V. Clinical practices in neonatal oxygenation: where have we failed? What can we do? J Perinatol. 2008; 28: S28-S34.
- Mauer M, Zinman B, Gardiner R, Suissa S, Sinaiko A, Strand T, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med. 2009; 361: 40-51.
- Sjølie AK, Klein R, Porta M, Orchard T, Fuller J, Parving HH, et al. Retinal microaneurysm count predicts progression and regression of diabetic retinopathy. Post-hoc results from the DIRECT Programme. Diabet Med. 2011; 28: 345-351.
- De Bundel D, Smolders I, Yang R, Albiston AL, Michotte Y, Chai SY. Angiotensin IV and LVV-haemorphin 7 enhance spatial sorking memory in rats: Effects on hippocampal glucose levels and blood flow. Neurobiol Learn Mem. 2009; 92: 19-26.
- Mountford SJ, Albiston AL, Charman WN, Ng L, Holien JK, Parker MW, et al. Synthesis, structure-activity relationships and brain uptake of a novel series of benzopyran inhibitors of insulin-regulated aminopeptidase. J Med Chem. 2014; 57: 1368-1377.
- Pham V, Albiston AL, Downes CE, Wong CH, Diwakarla S, Ng L, et al. Insulin-regulated aminopeptidase deficiency provides protection against ischemic stroke in mice. J Neurotrauma. 2012; 29: 1243-1248.
- Muthalif MM, Benter IF, Uddin MR, Harper JL, Malik KU. Signal transduction mechanisms involved in angiotensin-91-7)-stimulated arachidonic acid release and prostanoid synthesis in rabbit aortic smooth muscle cells. J Pharmacol Exp Ther. 1998; 284: 388-398.
- Ueda S, Masumori-Maemoto S, Ashino K, Nagohara T, Gotoh E, Umemura S, et al. Angiotensin (1-7) attenuates vasoconstriction evoked by angiotensin II but not by noradrenaline in man. Hypertension. 2000; 35: 998-1001.
- Loot AE, Roks AJ, Henning RH, Tio RA, Suurmeiher AJ, Boomsma F, et al. Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation. 2002; 105: 1548-1550.
- Campagnole-Santos MJ, Heringer SB, Batista EN, Khosla MC, Santos RA. Differential baroreceptor reflex modulation by centrally infused angiotensin peptides. Am J Physiol. 1992; 263: R89-R94.
- Silva-Barcellos NM, Frezard F, Caligiome S, Santos RA. Long-lasting cardiovascular effects of lipsome-kentrapped angiotensin-(1-7) at the rostral ventrolateral medulla. Hypertension. 2001; 38: 1266-1271.
- Feener EP, Northrup JM, Aieollo LP, King GL. Angiotensin II induces plasminogen activator inhibitor-1 and -2 expression in vascular endothelial and smooth muscle cells. J Clin Invest. 1995; 95: 1353-1362.
- Vaughan DE, Lazos SA, Tong K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest. 1995; 95: 995-1001.
- Kucharewicz I, Chabielska E, Pawlak D, Matys T, Rolkowski R, Buczko W. The antithrombotic effect of angiotensin (1-7) closely resembles that of losartan. J Renin Angiotensin Aldosterone Syst. 2000; 1: 268-272.
- Kucharewicz I, Pawlak R, Matys T, Chabielska E, Buczko W. Angiotensin (1- 7): An active member of the renin-angiotensin system. J Physiol Pharmacol. 2002; 53: 533-540.
- Klein N, Gembardt F, Supe S, Kaestle SM, Nickles H, Erfinanda L, et al. Angiotensin-(1-7) protects from experimental acute lung injury. Crit Care Med. 2013; 41: e334-e343.
- Lu B, Nagappan G, Lu Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol. 2014; 220: 220-223.
- Meldolesi J. Neurotrophin receptors in the pathogenesis, diagnosis and therapy of neurodegenerative diseases. Pharmacol Res. 2017; 121: 129- 137.
- Valenza M, Facchinetti R, Menegoni G, Steardo L, Scuderi C. Alternative targets to fight Alzheimer’s disease: Focus on astrocytes. Biomolecules. 2021; 11: 600.
- Boyce VS, Mendell LM. Neurotrophins and spinal circuit function. Front Neural Circuits. 2014; 8: 59.
- West AF, Prunslid P, Timmusk T. Neurotrophins: Transcription and translation. Handb Exp Pharmacol. 2014; 220: 67-100.
- Gibon J, Barker PA. Neurotrophins and proneuotrophins: Focus on synaptic activity and plasticity in the brain. Neuroscientist. 2017; 23: 587-604.
- Karatas H, Hemisci M, Eren-Kocak E, Dalkara T. Brain peptides for the treatment of neuropsychiatric disorders. Curr Pharm Des. 2018; 24: 3905- 3917.
- Wang R, Holsinger RMD. Exercise-induced brain-derived neurotrophic factor expression: Therapeutic implications for Alzheimer’s dementia. Aging Res Rev. 2018; 48: 109-121.
- O’Leary PD, Hughes RA. Design of potent peptide mimetics of brain-derived neurotrophic factor. J Biol Chem. 2003; 278: 25738-25744.
- Jang SW, Liu X, Yepes M, Shephard KR, Miller GW, Liu Y, et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci USA. 2010; 107: 2687-2692.
- Jang SW, Liu X, Chan CB, Weinshenker D, Hall RA, Xiao G, et al. Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkB heterodimerization and has potent neurotrophic activity. Chem Biol. 2009; 16: 644-665.
- Immunotherapy Study for Patients with Stage IV Melanoma. 2021.
- Royea J, Zhang L, Tong XK, Hamel E. Angiotensin IV receptors mediate the cognitive and cerebrovascular benefits of losartan in a mouse model of Alzheimer’s disease. J Neurosci. 2017; 37: 5562-5573.
- Sardinia MF, Hanesworth JM, Krebs LT, Harding JW. AT4 receptor binding characteristics: D-amino acid- and glycine-substituted peptides. Peptides. 1993; 14: 949-954.
- Sardinia MF, Hanesworth JM, Krishnan F, Harding JW. AT4 receptor structure-binding relationship: N-terminal-modified angiotensin IV analogues. Peptides. 1994; 15: 1399-1406.
- Krebs LT, Kramar EA, Hanesworth JM, Sardinia MF, Ball AE, Wright JW, et al. Characterization of the binding properties and physiological action of divalinal-angiotensin IV, a putative AT4 receptor antagonist. Regul Pept. 1996; 67: 123-130.
- Krebs LT, Kramar EA, Hanesworth JM, Sardinia MF, Ball AE, Wright JW, et al. Characterization of the binding properties and physiological action of divalinal-angiotensin IV, a putative AT4 receptor antagonist. Regul Pept. 1996; 67: 123-130.
- Kramár EA, Armstrong DL, Ikeda S, Wayner MJ, Harding JW, Wright JW. The effects of angiotensin IV analogs on long-term potentiation within the CA1 region of the hippocampus in vitro. Brain Res. 2001; 897: 114-121.