
Editorial
J Hepat Res. 2014;1(1): 1003.
New Findings about Liver Kupffer Cells/Macrophages, B Cells and their Functions
Shuhji Seki*, Masami Ikarashi, Manabu Kinoshita, Masahiro Nakashima and Hiroyuki Nakashima
Department of Immunology and Microbiology, National Defense Medical College, Japan
*Corresponding author: Shuhji Seki, Department of Immunology and Microbiology, National Defense Medical College, 3-2 Namiki, Tokorozawa, Saitama, Japan
Received: Aug 05, 2014; Accepted: Aug 05, 2014; Published: Aug 07, 2014
Editorial
Recent studies in our laboratory have elucidated important and interesting aspects of the innate immune cells in the liver. We found that mouse liver macrophages, F4/80+Kupffer cells, consist of three subsets; CD11b+ cells (30%), CD32+ cells (50%) and CD68+ cells (40%). Approximately one-half of CD68+ cells co-express CD32. These CD32+ cells are precursors of CD68+ cells, because a large proportion of CD32+ cells express molecules of immature precursors or stem cells, such as c-kit (CD117) and CD34 (Figure 1). Some CD32+CD68+ cells express these immature cell markers, while CD68+ cells have lost their expression of these markers [1,2]. Although CD32/68 cells predominate in the liver, they are rarely seen in the blood and spleen.
Mature liver CD68+ cells phagocytose bacteria (such as E. coli and S. aureus) and efficiently kill them in phagolysosomes by producing Reactive Oxygen Spices (ROS), but they produce only a small amount of cytokines (TNF, IL-12) [1,2]. On the other hand, CD11b+ cells have potent cytokine-producing activity (TNF, IL- 12), but they cannot effectively kill bacteria [1,2] (Figure 1). These cells are able to phagocytose bacteria, but they cannot produce functional phagolysosomes after the engulfment of the bacteria [1,2]. Consistently, CD32/68 cells were found to express a phagocytosisrelated complement receptor, CRIg, whereas CD11b+ cells did not [2] (Figure 1).
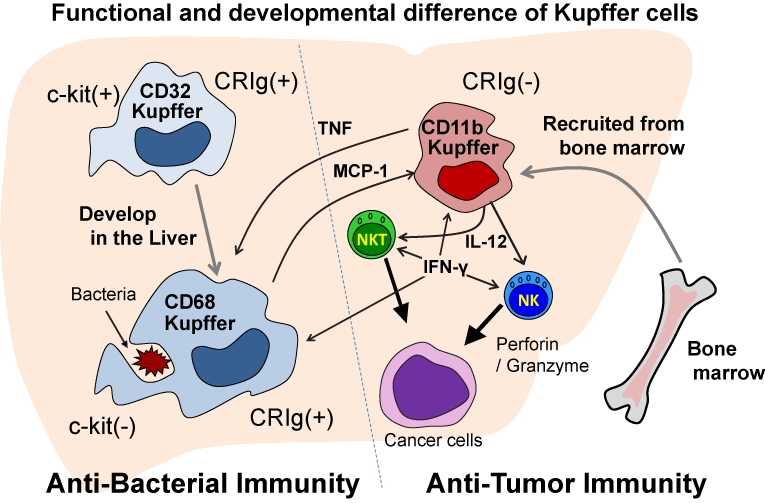
Figure 1: CD68 Kupffer cells develop from c-kit positive CD32 precursors
in the liver and have a potent phagocytic and bactericidal activity but do not
produce IL-12 nor TNF. CD11b Kupffer/macrophages are recruited to the liver
from bone marrow by MCP-1 produced by CD68 Kupffer cells and produce IL-
12 and TNF in response to tumors and bacteria, and IL-12 induces antitumor
activity by NK/NKT cells and IL-12/TNF induce inflammation. However,
CD11b Kupffer/macrophages cannot effectively kill bacteria due to inability to
form functional phagolysosomes.
A flow cytometric analysis and immunohistochemical studies revealed that the CD32/68 cells are large and spindle-shaped, while the CD11b cells are small and round or oval-shaped [2]. CD32/68 cells cannot be obtained unless the liver is treated with collagenase, while CD11b cells can be harvested without collagenase treatment [2]. Furthermore, CD32/68 cells are radio-resistant, while CD11b cells are extremely radio-sensitive. When CD11b cells were depleted in the liver, spleen and peripheral blood by 6 Gy irradiation, they could be reconstituted by bone marrow transplantation [2]. Conversely, clodronate-liposome (clod-lip) or GdCl3 injection into mice depleted the CD32/68 cells, but increased the CD11b cells in the liver, presumably because CD32/68 cells phagocytose toxic reagents and are frequently exposed to these toxic compounds, while CD11b cells may not be exposed to toxic compounds by their poor phagolytic activity [2].
Furthermore, we found that CD32/68 cells produce MCP-1 after they phagocytose clod-lip and before undergoing apoptosis, and MCP-1 leads to the accumulation of CD11b cells in the liver from the periphery and bone marrow, and probably stimulates their functions (TNF/IL-12 production) [2]. Therefore, mice depleted of CD32/68 cells in the liver by clod-lip administration became extremely susceptible to bacterial infections, whereas these mice unexpectedly became resistant to liver metastatic tumors, and lived longer after intra-splenic EL-4 tumor cell injection than control mice, because the accumulated and activated CD11b Kupffer cells/macrophages produced IL-12, which stimulates liver NK cells and NKT cells to produce IFN-? and may augment the liver's antitumor immunity [2] (Figure 1). CD32/68 cells, but not CD11b cells, express other markers specific for tissue macrophages, such as MerTK and CD64, which are also expressed by spleen CD11b-(minus) resident macrophages [2]. Thus, the mature CD68 cells in the liver are resident macrophages (Kupffer cells), while the F4/80+CD11b cells in the liver are recruited Kupffer cells/macrophages from bone marrow, and these cells are quite different from each other in terms of the their functions and development [2]. These features of liver Kupffer cells (the subsets and their functions)also apply to human liver Kupffer cells [2].
The concept that had been believed for long time that the same macrophages engulf bacteria and kill them by ROS and lytic enzymes and subsequently produce inflammatory cytokines needs to be reconsidered. Instead, F4/80+CD68+ resident macrophages in the liver and presumably F4/80+CD11b-cells in other organs (spleen, lungs, peritoneal cavity, etc.), phagocytose bacteria and kill them by ROS and lytic enzymes, namely anti-bacterial immunity. Recruited F4/80+CD11b+ macrophages in the liver and other organs produce IL-12 and TNF when they recognize bacteria, tumors and their antigens via their receptors, including Toll-like-receptors and CD1d, and then evoke antitumor immunity and inflammation. IL-12 activates NK cells and NKT cells to produce IFN-? and antitumor immunity, and IFN-? activates CD68 Kupffer cells to further augment their function in a positive feedback loop. TNF is also required for CD68 Kupffer cells to produce ROS [3] (Figure 1). Thus, both types of macrophages are activated and required for optimum immunity (Figure 1).
We have also made several interesting discoveries regarding mouse liver B cells. In our previous study, it was found that mouse liver B cells produce little IgM in response to LPS, but produce a substantial amount of IFN-? and IL-12 [4]. Such functions were quite different from those of spleen B cells, which produce a large amount of IgM following LPS stimulation, but do not produce any IFN-? or IL-12 [4]. Furthermore, LPS-binding B cells stimulate NK cells to produce IFN-? not only by their production of IL-12, but also by their direct contact with NK cells [4]. Since 35-40% of liver lymphocytes are B cells [4], B cells are a largest population of liver lymphocytes, and comprise a larger proportion of cells than NK cells, NKT cells or T cells in the liver. Therefore, B cells may play a much more important role than previously thought in gram negative bacterial infections and in the T helper 1 (Th1) immune response.
Since IL-12 is a representative Th1 cytokine produced by macrophages, the IL-12 production of liver B cells led us to hypothesize that liver B cells may have phagocytic activity, and this was found to be the case (Figure 2). Both B-1 B cells and B-2 B cells in the liver could engulf not only latex beads, but also living E. coli [5,6]. Furthermore, a killing assay including CFU assays confirmed that liver B cells killed E. coli. By functional phagolysosomes, which was also confirmed by pHrodo-conjugated E. coli, LysoTracker is staining and acid-phosphatase staining. Spleen B cells also exhibited phagocytosis of E. coli, but they could not kill the bacteria because they could not form functional phagolysosomes [5]. In addition, the killing activity of liver B cells was increased by mouse fresh serum, but heat-inactivation of the serum abrogated this enhancement, suggesting that B cell phagocytosis involves opsonization [5]. Parra et al. also reported that mouse peritoneal B-1 cells also phagocytosed latex beads and phagocytosed and killed E. coli [7]. These findings in B cells invite us to revisit the intersection of Mechnikov's theory (the phagocytosis for bacterial killing) and Erlich's theory (antibodies for bacteria killing), which were given the Nobel Prize for together in 1908 [6]. The B cells thus play an important role in both innate immunity (phagocytosis) and adaptive immunity (antibody production). Further studies are necessary to clarify the relationship between phagocytosis and IgM production of B cells.
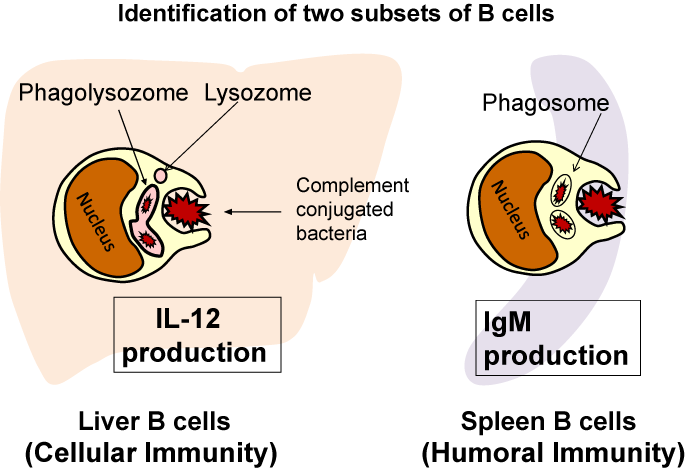
Figure 2: Liver B cells phagocytose E. coli, kill them in phagolysosomes and
produce IL-12 but produce little amount of IgM in response to E. coli and LPS,
while spleen B cells produce a large amount of IgM in response to E. coli and
LPS but produce little amount of IL-12 and do not kill E. coli because they
cannot form phagolysosomes.
From the viewpoint of the two different subsets of Kupffer cells, not only antitumor immunity and antibacterial infection immunity, but also the mechanisms underlying various experimental hepatitis, become clearer. We previously reported that the NKT cells activated by TNF expressed FasL and induced hepatocyte apoptosis in a model of acute hepatitis induced in mice by a-galactosylceramide, a specific ligand of NKT cells [8-10]. This is also the case for acute hepatitis induced by bacterial DNA (CpG-ODN) [11]. Now, it is obvious that the TNF in these cases of hepatitis is produced by recruited CD11b macrophages/Kupffer cells [12]. Furthermore, both hepatitis were aggravated when mice were fed with high fat and high cholesterol diet, because liver CD11b macrophages/Kupffer cells increased and produce several-fold greater amount of TNF than those of control diet mice, which can be a mechanism of Non-Alcoholic Steatohepatitis (NASH) [12]. It was also previously reported by our group that Concanavalin-A (Con-A) hepatitis in mice was induced by the ROS produced by CD68 Kupffer cells. Although this was found before the publication of our report dealing with Kupffer cell subsets [1], we noted at that time that there might have been two different types of Kupffer cells, because while GdCl3 depleted only a part of the Kupffer cells, it completely inhibited Con-A hepatitis, but with no effect on the serum TNF levels [3]. Therefore, we started to consider the possibility that different types of Kupffer cells produced ROS and TNF. However, an anti-TNF Ab also completely inhibited Con-A hepatitis, because the TNF produced by recruited CD11b macrophages/Kupffer cells is required for the activation and ROS production of resident CD68 Kupffer cells to induce hepatic injury but increased TNF itself cannot cause hepatic injury [3]. The depletion of NKT cells also greatly improved Con-A hepatitis; however, the NKT cells or their FasL are not the final effectors, although NKT cells are required for the increase in CD68 Kupffer cells [3]. Thus, without consideration of tow subsets of Kupffer cells (Figure 1), the mechanism of Con-hepatitis is hard to grasp.
Finally, we introduce the immune mechanism underlying the acute hepatic injury induced by CCl4, which we have recently explored [13]. Although CCl4-induced hepatitis is a well-known toxic chemical-induced hepatitis, the immune mechanism involved in the acute phase of this hepatitis has not been extensively examined. At 12 h after CCl4 injection into mice, up to 20% of the liver CD11b Kupffer cells/macrophages express intracellular TNF and surface FasL, before the hepatic injury peaks at 24h [13]. Furthermore, the depletion of CD68 Kupffer cells by clod-lip pretreatment markedly aggravated the hepatic injury by increasing the CD11b Kupffer cells/macrophages, which was associated with increased serum TNF levels. Either anti- TNF antibody or anti-FasL Ab pretreatment improved hepatic injury, which was also found in FasL-deficient gld mice. However, hepatic injury was not attenuated in mice depleted of either NKT cells (genetically or via antibody treatment) or NK cells, suggesting that FasL-expressing CD11b Kupffer cells are hepatotoxic effectors [13]. Furthermore, the adoptive transfer of CD11b Kupffer cells and in vitro cytotoxic assays using primary cultured hepatocytes confirmed that CD11b Kupffer cells are indeed final effectors [13].
Interestingly, in the hepatitis induced by either a-galactosylceramide, CpG-ODN or Con-A, the plasma TNF levels peaked around 1 h after the injection of reagents, while the TNF levels peaked at 24 h after CCl4 injection [13]. Therefore, CD11b Kupffer cells more slowly produce TNF in CCl4 hepatitis than in other types of hepatitis, suggesting that the effectors in other types of hepatitis, namely FasL-expressing NKT cells or ROS-producing CD68 Kupffer cells, may directly attack hepatocytes, while FasL-expressing CD11b Kupffer cells in CCl4 hepatitis may slowly attack the hepatocytes after they were injured by CCl4.
Adopting our concepts of liver Kupffer cells and B cells (Figure 1,2), new prospects may develop with regard to the understanding of various pathophysiological conditions and diseases of the liver.
References
- Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, et al. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol. 2010; 53: 903-910.
- Ikarashi M, Nakashima H, Kinoshita M, Sato A, Nakashima M, Miyazaki H, et al. Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. J Leukoc Biol. 2013; 94: 1325-1336.
- Nakashima H, Kinoshita M, Nakashima M, Habu Y, Shono S, Uchida T, et al. Superoxide produced by Kupffer cells is an essential effector in concanavalin A-induced hepatitis in mice. Hepatology. 2008; 48: 1979-1988.
- Matsumoto A, Kinoshita M, Ono S, Tsujimoto H, Majima T, Habu Y, et al. Cooperative IFN-gamma production of mouse liver B cells and natural killer cells stimulated with lipopolysaccharide. J Hepatol. 2006; 45: 290-298.
- Nakashima M, Kinoshita M, Nakashima H, Habu Y, Miyazaki H, Shono S, et al. Pivotal advance: characterization of mouse liver phagocytic B cells in innate immunity. J Leukoc Biol. 2012; 91: 537-546.
- Cancro MP. Editorial: phagocytic B cells: déjá vu all over again? J Leukoc Biol. 2012; 91: 519-521.
- Parra D, Rieger AM, Li J, Zhang YA, Randall LM, Hunter CA, et al. Pivotal advance: peritoneal cavity B-1 B cells have phagocytic and microbicidal capacities and present phagocytosed antigen to CD4+ T cells. J Leukoc Biol. 2012; 91: 525-536.
- Inui T, Nakashima H, Habu Y, Nakagawa R, Fukasawa M, Kinoshita M, et al. Neutralization of tumor necrosis factor abrogates hepatic failure induced by alpha-galactosylceramide without attenuating its antitumor effect in aged mice. J Hepatol. 2005; 43: 670-678.
- Inui T, Nakagawa R, Ohkura S, Habu Y, Koike Y, Motoki K, et al. Age-associated augmentation of the synthetic ligand- mediated function of mouse NK1.1 ag(+) T cells: their cytokine production and hepatotoxicity in vivo and in vitro. J Immunol. 2002; 169: 6127-6132.
- Nakagawa R, Nagafune I, Tazunoki Y, Ehara H, Tomura H, Iijima R, et al. Mechanisms of the antimetastatic effect in the liver and of the hepatocyte injury induced by alpha-galactosylceramide in mice. J Immunol. 2001; 166: 6578-6584.
- Kawabata T, Kinoshita M, Inatsu A, Habu Y, Nakashima H, Shinomiya N, et al. Functional alterations of liver innate immunity of mice with aging in response to CpG-oligodeoxynucleotide. Hepatology. 2008; 48: 1586-1597.
- Nakashima H, Ogawa Y, Shono S, Kinoshita M, Nakashima M, Sato A, et al. Activation of CD11b+ Kupffer cells/macrophages as a common cause for exacerbation of TNF/Fas-ligand-dependent hepatitis in hypercholesterolemic mice. PLoS One. 2013; 8: e49339.
- Sato A, Nakashima H, Nakashima M, Ikarashi M, Nishiyama K, Kinoshita M, et al. Involvement of the TNF and FasL produced by CD11b Kupffer cells/macrophages in CCl4-induced acute hepatic injury. PLoS One. 2014; 9: e92515.