
Review Article
J Immun Res. 2014;1(2): 7.
Hypoxia: A Key Feature of the Tumor Microenvironment Triggers Several Mechanisms of Evasion from Natural Killer and Cytotoxic T Lymphocytes Surveillance
Mgrditchian T1, Arakelian T1, Paggetti J1, Viry E1, Al-Absi A2, Medves S1, Moussay E1, Berchem G1,3, Thomas C2 and Janji B1*
11Laboratory of Experimental Hemato-Oncology, Department of Oncology, Public Research Center for Health
2Laboratory of Cellular and Molecular-Oncology, Department of Oncology, Public Research Center for Health, Luxembourg
3Centre Hospitalier de Luxembourg, Department of Hemato-Oncology, Luxembourg
*Corresponding author: Bassam Janji, Laboratory of Experimental Hemato-Oncology, Department of Oncology, Public Research Center for Health, Luxembourg
Received: November 04, 2014; Accepted: November 18, 2014; Published: November 20, 2014
Abstract
Since many years, the tumor microenvironment is recognized as an important promoter of cancer initiation and progression. Although tumors primarily consist in cancer cells, various components/factors of the microenvironment and more specifically the immune landscape, dramatically impact cancer progression. Comprehensive analyses conducted on diverse tumors have identified and characterized the most relevant components/ factors of the tumor microenvironment that support the malignant behaviorof a growing primary tumor. Tumor hypoxia is a common characteristic of the tumor microenvironment that is associated with tumor progression, metastasis, treatment failure and escape from immune surveillance. Although immune cells are usually efficiently recruited into the tumor bed, hypoxic microenvironment was reported to compromise immune cell functions and, in some cases, switch immune cell activity towards a pro-tumorigenic profile. Mechanistic studies have highlighted that hypoxia acts as double-edged sword: it simultaneously impairs the function of immune cells in the tumor microenvironment and activates intrinsic cell resistance mechanisms in tumor cells. The present article aims at reviewing some recent findings on how hypoxia impairs the anti-tumor immune response by focusing on emerging mechanisms by which hypoxia damps the function of immune effectors cells and activates intrinsic immune resistance mechanisms in tumor cells including autophagy and actin cytoskeleton remodeling.
Keywords: Cancer; Hypoxia; Immune response; Tumor microenvironment; Autophagy; Actin cytoskeleton
Abbreviations
AFs: Actin Filaments; APCs: Antigen Presenting Cells; ARP: Actin-Related Protein; ATG: Autophagy-Related Genes; Bcl-2: B Cell Lymphoma 2; CAFs: Cancer-Associated Fibroblasts; Cdc42: Cell Division Cycle 42; CTL: Cytotoxic T Lymphocyte; ECM: Extracellular Matrix; EMT: Epithelial-to-Mesenchymal Transition; FAK: Focal Adhesion Kinase; HIF: Hypoxia-Inducible Factors; HOXA1: Homeobox A1; HRE: Hypoxia-Response Element; IS, Immunological Synapse; LFA: Lymphocyte Function-Associated Antigen; MAPK: Mitogen-Activated Protein Kinases; MHC: Major Histocompatibility Complex; MICA: MHC Class I Polypeptide-Related Sequence A; miR: micro RNA; NK: Natural Killer; NKG2D: Natural Killer Group 2 Member D; PHD2: Prolyl Hydroxylase Domain Protein 2; PTPN1: Protein Tyrosine Phosphatase Non-Receptor Type 1; Rac1: Ras-Related C3 Botulinum Toxin Substrate 1; Rho: Ras Homolog; ROCK1: Rho-Associated Protein Kinase 1; STAT3: Signal Transducer and Activator of Transcription 3; TCR: T Cell Receptor; TNF: Tumor Necrosis Factor; TP53I11: Tumor Protein p53-inducible Protein 11; VEGF: Vascular Endothelial Growth Factor; VHL: Von Hippel– Lindau; WAS: Wiskott-Aldrich Syndrome; WASp: WAS Protein; YAP: Yes-associated Protein.
Introduction
Research in tumor immunology has validated the concept of cancer immune surveillance which predicts that the immune system can recognize precursors of cancer and, in most cases, destroy them or slow their growth before they become clinically apparent [1, 2]. Several types of immune cells are involved in tumor immune surveillance. Briefly, key cells of the adaptive immune system recognizing cancer cells are cytotoxic T lymphocytes (CTL) which are able to identify tumor antigens via the T cell receptor (TCR) [3]. Some of these antigens are expressed exclusively by tumors and thus are called tumor-specific antigens [3]. Natural killer (NK) cells of the innate immune system also play an important role in tumor immune surveillance [3] by mechanisms called “missing-self” and “induced-self” recognitions [5]. In addition to CTL and NK cells, macrophages and neutrophil granulocytes are also involved in antitumor immunity [6]. Macrophages are antigen presenting cells (APCs) that display tumor antigens and stimulate other immune cells such as CTL, NK cells and other APCs [7]. While the molecular mechanism by which CTL and NK cells recognize their target tumor cells is fundamentally different, both immune cells kill their target following the establishment of immunological synapse (IS) [8]. The formation of IS requires cell polarization and extensive remodeling of the actin cytoskeleton at various stages [9]. It is now well established that CTL and NK cells recognize and kill target cells by two major pathways: either through the release of cytotoxic granules containing perforin and granzymes to the cytosol of target cells [10], or through tumor necrosis factor (TNF) super family-dependent killing [11].
Many microenvironmental factors (e.g. hypoxia, composition/ organization of the extracellular matrix (ECM), inflammation, immune suppressive tumor-associated cells) contribute to various aspects of cancer progression, including immune evasion of tumor cells [12, 13]. Recently, it has been reported that the immune system also sculpts the immunogenic phenotype of an established tumor to favor the emergence of resistant tumor cell variants [14]. Accumulating experimental and clinical evidence indicates that multiple mechanisms suppressing the anti-tumor immune functions are directly evolved in the tumor microenvironment [15]. Recently, attention has been focused on the mechanisms by which hypoxic stress within the tumor microenvironment alters tumor transcriptional profiles to modulate glycolysis, proliferation, survival and invasion [16].
In this review, we focus on recent progresses in understanding the influence of hypoxic stress on the tumor survival mechanisms and tumor escape from immune surveillance. We discuss how hypoxia impairs tumor cell killing mediated by both innate and adaptive immune cells. We also review how hypoxia confers resistance to immune attack by activating intrinsic resistance mechanisms in tumor cells. Particular attention is given to hypoxia-induced immune escape mechanisms that rely on actin cytoskeleton remodeling in tumor or immune cells. Finally, we briefly address how hypoxia-targeted strategies may have potential clinical application for restoring an efficient anti-tumor immune response.
Hypoxia and Hypoxia-inducible Factors (HIF) Regulation
Hypoxic stress plays a major role in the acquisition of tumor resistance to immune cell-mediated apoptosis and in the impairment of immune cell activity. Tumor cells adapt to hypoxic microenvironment by inducing transcription factors of the hypoxia inducible factor (HIF) family [17]. The HIF family comprises three members, HIF-1,-2 and-3. HIF-1 is a hetero dimeric protein composed of a constitutively expressed HIF-1β subunit and an oxygen-regulated HIF-1α subunit. In the presence of oxygen, HIF-1α is hydroxylated on proline residue 402 and/or 564 by prolyl hydroxylase domain protein 2 (PHD2). Hydroxylated HIF-1α interacts with the Von Hippel–Lindau (VHL) tumor suppressor protein [18] and recruits an E3 ubiquitin-protein ligase to catalyze the polyubiquitination of HIF-1α and its subsequent targeting to the ubiquitin proteasome system for degradation [19] (Figure 1). Under hypoxic condition, the hydroxylation of HIF-1α is inhibited leading to HIF-1α accumulation. Subsequently, HIF-1α translocates to the nucleus, dimerizes with HIF-1β subunit, binds to the hypoxia-response elements (HRE) present in the promoter of target genes, and recruits co-activators to activate gene transcription (Figure 1). Similar to HIF-1α, HIF-2α is regulated by oxygen-dependent hydroxylation [20]. Although they share structurally similar DNA-binding and dimerization domains,HIF-1α and -2α differ in their transactivation domains. Accordingly, HIF-1α and -2α share overlapping target genes, and each also regulates a set of specific targets [21]. HIF-3α lacks the transactivation domain and was proposed to function as an inhibitor of HIF-1α and HIF-2α. Interestingly, its expression is transcriptionally regulated by HIF-1[22].
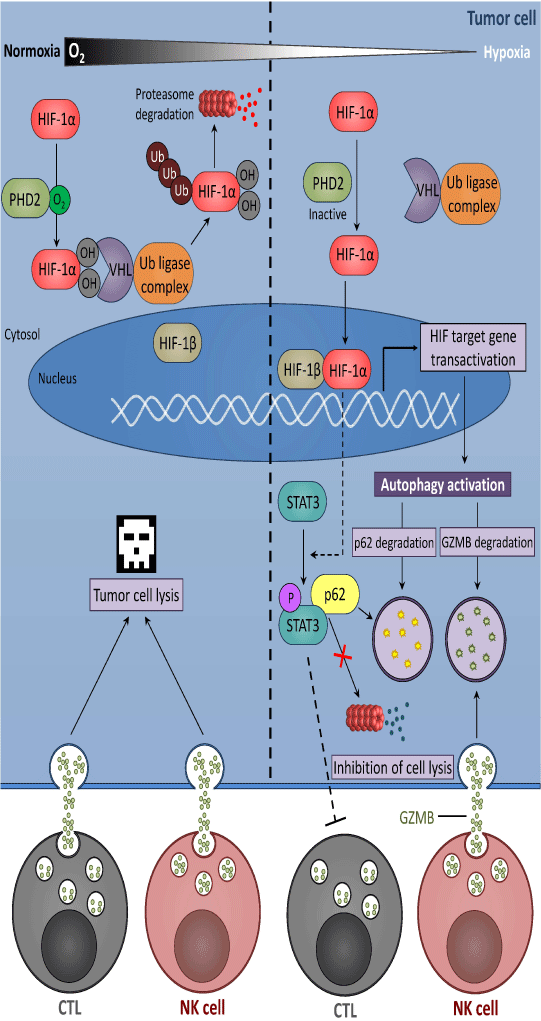
Figure 1: Hypoxia induces tumor cell resistance mechanisms leading to escape from CTL- or NK-mediated lysis. Under normoxic condition, the oxygen-sensitive prolyl hydroxylase domain protein 2 (PHD2) hydroxylatesHIF-1a subunit which is bound to Von Hippel-Lindau protein (VHL), ubiquitinated and subsequently degraded by the ubiquitin-proteasome system. Under hypoxia, PHD2 protein is inactivated, and HIF-1a is therefore stabilized. Stabilized HIF-1a translocated to the nucleus and formed with HIF-1β, a heterodimeric complex responsible for transcriptional activation of HIF-target genes including those responsible for the activation of autophagy. In addition, HIF induced the phosphorylation of the signal transducer and activator of transcription (STAT) 3 by a mechanism which is not fully described. The activation of autophagy leads to the degradation of the cargo protein p62, responsible for the elimination of phospho-STAT3 by the ubiquitin proteasome system. The degradation of phospho-STAT3 inhibitsCTL-mediated lysis of tumor cells. Furthermore, excessive autophagy in hypoxic target cells leads to the selective degradation of granzyme B (GZMB), a serine protease contained into the cytotoxic granules released by natural killer (NK) cells, thereby inhibiting NK-mediated lysis.
Influence of Hypoxia on Cancer Cell Sensitivity to CTL and NK-mediated Lysis
As previously mentioned, hypoxia modulates both the activity of immune effectors and the response of tumor cells to these effectors. It has been reported that hypoxia decreases the sensitivity of tumor cells to CTL-mediated killing by several mechanisms involving HIF-1. For example, Noman et al. showed that HIF-1α induces the phosphorylation of signal transducer and activator of transcription 3 (STAT3) in tumor cells by a mechanism involving at least in part, vascular endothelial growth factor (VEGF) secretion. Following its translocation to the nucleus, HIF-1α cooperates with pSTAT3 to alter the susceptibility of non-small cell lung carcinoma to CTL-mediated killing [23] (Figure 1). A follow-up study has reported that the level of pSTAT3 is tightly controlled by the activation of autophagy in hypoxic cells. Indeed, the accumulation of pSTAT3 was no longer observed when the autophagic flux was inhibited in hypoxic tumor cells [24]. Briefly, autophagy is a catabolic process where a cell self-digests its own components. It can be activated in response to multiple stresses including hypoxia, nutrient starvation, growth factor withdrawal and endoplasmic reticulum stress. Autophagy primarily serves as an adaptive metabolic response providing nutrients, preventing accumulation of altered cell components [25], shaping the anti-tumor immune response [26] and the acquisition of resistance to TNF α [27]. Studies aimed at investigating the mechanisms by which autophagy controls pSTAT3 revealed that targeting autophagy decreases the level of pSTAT3, and that this decrease was mediated by the ubiquitin proteasome system [24, 28].
Another study showed that the transcription factor NANOG, which is involved in stem cell self-renewal, is a new mediator of hypoxia-induced resistance to CTL-mediated lysis [29, 30]. NANOG is induced at both transcriptional and protein levels under hypoxic stress and its targeting leads to significant attenuation of hypoxiainduced tumor resistance to CTL-dependent killing. NANOG depletion results in inhibition of STAT3 phosphorylation and nuclear translocation. Furthermore, it has been shown that HIF-1 induces hypoxia-inducible micro RNA (miR)-210. Overexpression of miR-210 in tumor cells targets protein tyrosine phosphatase non-receptor type 1 (PTPN1), homeobox A1 (HOXA1), and tumor protein p53-inducible protein 11 (TP53I11), and thereby decreases tumor cell susceptibility to CTL [31]. Hypoxia has also been reported to increase the shedding of the major histocompatibility complex (MHC) class I polypeptide-related sequence A (MICA), a ligand for the activating receptor natural killer group 2 member D (NKG2D), at the surface of prostate cancer cells. This shedding is related to the impairment of NO signaling [32] and leads to tumor escape from NK and CTL cells. Furthermore, HIF-1 is able to downregulate MICA expression in osteosarcoma cells resulting in tumor resistance to NKmediated lysis [33]. Recently, we showed that targeting autophagy under hypoxia restores NK-mediated lysis in breast cancer cells. We provided mechanistic evidence that activation of autophagy under hypoxia leads to the degradation of NK-derived granzyme B making hypoxic tumor cells less sensitive to NK-mediated killing [34, 35] (Figure 1). Taken together, it is now well documented that hypoxic microenvironment is an important determinant involved in the control of anti-tumor immune response. Indeed, its immune suppressive effect contributes not only in damping the immune cell function but also in conferring resistance mechanisms to tumor cells to resist immune cells attack [36].
The Actin Cytoskeleton: A Point of Convergence for Various Processes Involved in Tumor Immune Evasion
To fulfill their biological functions, immune cells require exceptional capabilities to proliferate, move through the entire body, infiltrate tissues, establish cell-cell contacts and exchange various types of compounds/signals with both other immune cells and their targets. The related cellular processes, including cell division, cell migration and invasion, cell shape plasticity, intracellular transport,endocytosis and exocytosis, membrane trafficking and phagocytosis, largely rely on dynamic re-arrangements of the actin cytoskeleton. Accordingly, dysregulation of the actin cytoskeleton organization or dynamics can compromise immune cell function and lead to severe diseases, including cancer [37]. A typical illustration is provided by the Wiskott-Aldrich syndrome (WAS) which is caused by alterations of WAS protein (WASp) activity. One major function of WASp is to stimulate actin polymerization through the actin-related proteins (ARP) 2/3 nucleation complex in response to signals from the cell surface [38, 39]. Although some signaling activities of WASp are independent of actin remodeling, most immune cell defects induced by the loss of WASp are at least to some extent, related to defective actin responses, including cell migration, activation, IS formation, and phagocytosis [40-43].
One important aspect of the anti-tumor immune response is the establishment of the IS between cytotoxic immune cells, such as CTL and NK cells, and their targets. Although the signaling pathways activating CTL and NK cell-mediated tumor cell death differ, the establishment of a functional IS and the directed secretion of cytolytic granules toward the tumor cell are relatively well conserved processes that require extensive cytoskeletal remodeling. Following immune cell activation, actin polymerization is activated [44, 45] and granules polarize toward the cell-cell contact region in a microtubule network and microtubule motor activity-dependent manner [46-48]. A critical feature of this process is the translocation of the microtubuleorganizing center (upon which granules converge) from the rear of the immune cell to the leading edge where the immunological synapse is established. The presence of pre-docked granules at the cell cortex of CTL and NK cells, that can be exocytosed before centrosome polarization has been reported [49-51]. Both pharmacological and genetic studies indicate that immune cell activation, IS formation, and the steps following granule polarization largely rely on actin cytoskeleton remodeling [46, 48, 52-54]. A substantial remodeling event occurring simultaneously with centrosome translocation is the accumulation of actin toward the edge of the cell-cell contact region in the so-called distal supra molecular activation cluster where integrin proteins concentrate to mediate cell adhesion [55-57]. The central region of mature synapses was initially proposed to function as an actin-free clearance through which granules are passively released [47, 56]. Such a model was however challenged by studies supporting that granules associate with actin in a myosin IIA dependent manner prior to their delivery to the precise de-granulation sites [58, 59]. Recently, high-resolution microscopy has provided compelling evidence that an actin meshwork is present at the center of the IS from NK cells and that it functions as a facilitator for granule secretion rather than as a barrier [46, 60-62].
In contrast to the well-characterized configurations and roles of actin filaments (AFs) in NK and CTL cells during the cytolytic process, there is a relative gap of knowledge about AF organization and dynamics in target tumor cells. However growing evidence indicate that AF reorganization in tumor cells can promote resistance to anti-tumor immunity (Figure 2). Thus, knock-down of two actinregulatory proteins, namely the actin-severing factor scinderin [63, 64] and the signaling molecule ephrin-A1 [65] that are overexpressed in resistant lung tumor cells, was sufficient to restore normal cell morphology and to increase cell sensitivity to CTL-mediated cell death [66]. A follow-up study established that the focal adhesion kinase (FAK) signaling pathway, which controls actin organization and modulates cell adhesion and proliferation was defective in tumor resistant cells [67]. Remarkably, the suppression of FAK in tumor cell was sufficient to promote their resistance to CTL by blocking IS formation. Furthermore, increased AF rearrangement and adhesion following pharmacological activation of Ras homolog (Rho) GTP ases has been reported to induce tumor cell susceptibility to CTL mediated killing. These data further support the role of actin remodeling as a switch in the control of tumor cell susceptibility to cytotoxic immune cells. In addition, depolymerization of AFs in NKsensitive target cells abrogates the conjugate formation mediated by lymphocyte associated antigen (LFA)-1 and subsequently affects granule polarization [68]. Experimental data point to a mechanism where actin cytoskeleton-dependent tethering of LFA-1 ligand such as intercellular adhesion molecules (ICAM)-1 and -2, is required for proper integrin signaling in NK cells [68]. Thus, the modulation of the lateral diffusion of integrin ligand by actin remodeling at the tumor cell cortex represents a potential facet of the immune evasion.
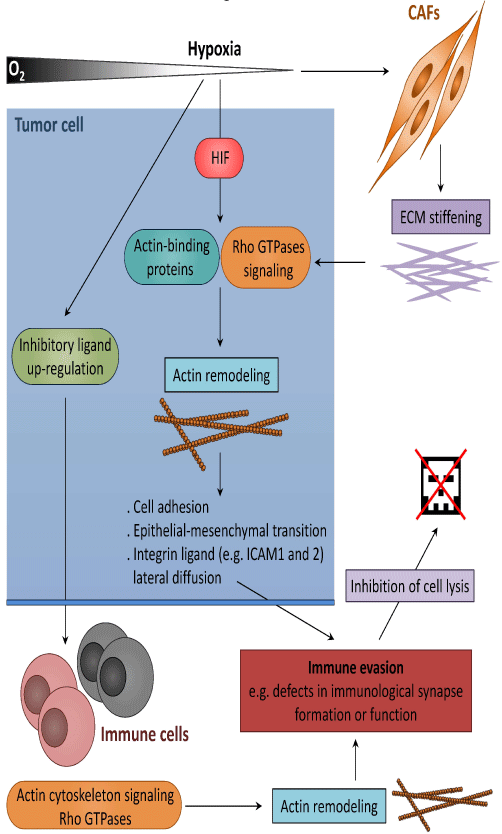
Figure 2: Hypoxia promotes tumor immune escape through actin cytoskeleton remodeling. Hypoxia can alter the expression of actin-binding proteins and/or Rho GTPase-mediated actin and adhesion signaling pathways either directly (in a HIF-dependent manner) or indirectly through extra cellular matrix (ECM) stiffening. The subsequent cytoskeletal changes modify cell adhesion, lateral diffusion of integrin ligands and promote epithelial-mesenchymal transition (EMT). This in turn drives tumor immune escape by compromising the formation of functional immunological synapses. In addition, the up regulation of inhibitory ligands at the surface of tumor cells compromise Rho GTPase activation signaling in immune cells, resulting in impaired cell migration and capacity to establish stable and functional immunological synapse.
Tumor cells do not only remodel their own actin cytoskeleton to escape from the immune system but are also capable of promoting cytoskeletal modifications in immune cells to impair their activity (Figure 2). Gene-expression profiling study revealed that genes involved in actin cytoskeleton formation and organization in peripheral blood CTL are considerably dysregulated in chronic lymphocytic leukemia patients [69]. Thus, leukemia cells were able to induce similar cytoskeletal defects in CTL from healthy allogeneic donors. Interestingly, the underlying process required cell-cell contact and was independent on cytokine release. Follow-up studies established that malignant cells up-regulate inhibitory ligands such as the B7 super family-related ligands CD200, CD274 and CD276, and TNF-receptor super family member CD270, to compromise the activation of key regulators of T cell synapse actin dynamics, including the Rho GTPases RhoA, Ras-related C3 botulinum toxin substrate 1 (Rac1) and cell division cycle protein 42 (Cdc42) and eventually compromise the establishment of a functional IS [70, 71]. Remarkably, similar inhibitory ligand signaling axes were found in a range of other hematologic and solid carcinoma cells, suggesting that tumor cell-induced T cell actin synapse dysfunction is a common immunosuppressive strategy. Recently, elegant work by Ramsay et al demonstrated that leukemia cells from patients impair T cell LFA- 1-mediated migration by altering Rho GTPase activation signaling. Importantly, the immune modulating drug lenalidomide was shown to restore T cell Rho GTPase activity and LFA-1 activation, laying the ground for immunotherapy approaches promoting T cell motility in cancer patients [72].
Tumor Microenvironment-induced Cancer Progression and Immune Evasion Involve Actin Cytoskeleton Remodeling
It has been reported that the physical properties of tumor stroma considerably differ from those of corresponding healthy tissues. Hypoxic stress within the tumor microenvironment induces an increase in ECM stiffness [73] and this increase is largely associated with the ability of hypoxia to induce the expression of ECM genes in cancer-associated fibroblasts, such as those involved in collagen fiber deposition and organization [74-76]. The perception of matrix stiffening by actin cytoskeleton-linked integrins activates signaling pathways that promote Rho GTPase-dependent cytoskeletal tension, focal adhesion assembly and cell migration [73, 77]. Interestingly, a recent study showed that an increase in matrix stiffness and actin cytoskeleton contractibility activates the transcription factor Yesassociated protein(YAP) in cancer-associated fibroblasts and that this activation is required for these fibroblasts to further increase matrix stiffening, cell invasion and angiogenesis [78]. Thus, by inducing a stiff microenvironment, hypoxia promotes actin remodeling in both tumor and cancer-associated fibroblasts (CAFs) leading to increased fibrillar collagen deposition and promoting tumorigenic functions of CAFs (Figure 2).
Hypoxic stress has been described to modify the expression and/ or activity of Rho GTPases and/or of their downstream targets in both healthy and cancer cells. Recently, HIF-1 and -2 were shown to directly activate transcription of RhoA and Rho-associated protein kinase 1 (ROCK1) genes, and subsequently promote actin cytoskeleton changes that underlie the invasive tumor cell phenotype [79]. Such coordination between HIF and RhoA-ROCK1 activation also induced FAK activation and focal adhesion formation, indicating that FAK is a major effectors of RhoA/ROCK1 in hypoxic breast cancer cells. Rac1 and Cdc42, two other major Rho GTPases controlling actin dynamics [80] and cancer progression [81] are upregulated and activated by hypoxia in different tumor cells [82, 83]. In this context, it is worth noting that an immune functional screen identified Cdc42 as a key mediator of tumor resistance to CTLmediated killing [84]. Remarkably, stable expression of constitutively active Cdc42 protected colorectal tumor cells against cytotoxicity and tumor suppression by alloreactive CTL in vitro and CTL and NK cells in vivo. However, the role of actin remodeling in the underlying mechanism remains uncertain. Instead, Cdc42 has been shown to promote immune resistance, at least in part, by mitogen-activated protein kinases (MAPK)-dependent inhibition of apoptosis and stabilization of the B cell lymphoma 2 (Bcl-2) antiapoptotic factor. In addition to modulating actin regulatory signaling pathways, hypoxia also drives changes in the expression level of specific actin-binding proteins [85]. However, how such changes influence the immune tumor response remains to be assessed.
As previously reported, hypoxia-induced autophagy plays important roles in tumor immune escape [86]. To our best knowledge, the role of the actin cytoskeleton in hypoxia-induced autophagy has not been extensively investigated in the specific context of tumor immune escape. Nevertheless, emerging evidence indicate that epithelial-tomesenchymal transition (EMT), a biological process associated with dramatic morphologic changes and actin cytoskeleton remodeling, alters the susceptibility of tumor cells to CTL-mediated immune surveillance by a mechanism involving autophagy [87, 88]. There is also clear evidence that Rho family-dependent actin remodeling is required for proper autophagy induced by other environmental stresses, such as starvation [89]. Finally, several autophagy-related genes (ATG) proteins were reported to physically and/or functionally interact with actin cytoskeleton components, such as the actin motor myosin II [90] and the FA protein paxillin [91].
Concluding Remarks
The ability of cancer cells to evade immune surveillance and resist immunotherapy raises fundamental questions about how tumor cells survive in the presence of a competent immune system. To address this issue, studies have primarily focused on the mechanisms by which tumor cells avoid recognition by the immune system without considering the impact of the tumor microenvironment. Thus, despite intense investigation, the relatively modest gains provided by immunotherapy can be in part attributed to the activation of mechanisms suppressing the anti-tumor immunity. It is now clearly established that the majority of these mechanisms are likely evolved in the local tumor microenvironment. In line with this, it may be more accurate to consider cancer, which was initially thought to be a disease of cells, then of genes, and then of genomes, as a disease of the microenvironment. While remarkable and fairly rapid progresses have been made over the past two decades regarding the role of the microenvironment in cancer biology and treatment, our understanding of its actual contribution in tumor resistance to immune cell attack is still fragmented.
Emerging data indicate that hypoxia in the tumor microenvironment plays key role in mediating tolerance to immune cell attack. Therefore, an understanding of the tumor microenvironment, and in particular of hypoxia-induced tumor resistance, may allow better understanding of tumor adaptation and evolution, and ultimately lead to improve the efficacy of potential therapeutics. Thus, it stands to reason that targeting hypoxia could have a substantial effect in evolving the properties of tumor microenvironment from immunosuppressive to immune permissive. In keeping with this, an extensive number of HIF inhibitory molecules are currently under development and being tested or in use in clinical settings. Such inhibitors comprise HIF-a antisense, dominant negative HIF-a and viral vectors as well as small molecules that inhibit HIF at multiple sites. Briefly, these inhibitors have been shown to block tumor xenograft growth and inhibit HIF activity through a wide variety of molecular mechanisms, including decreased HIF-1a mRNA levels, decreased HIF-1a protein synthesis, increased HIF-1a degradation, decreased HIF subunit hetero-dimerization, decreased HIF binding to DNA, and decreased HIF transcriptional activity. Notably, HIF-1a inhibitors would be expected to affect some parameters of the antitumor immunity such as CLT activity in vivo and in vitro (i.e., cytokine production, proliferation, and activation). Indeed, several recent studies highlight that hypoxia is capable of mediating tumor cell resistance to CTL and NK cells [24, 31, 36]. In addition, it should be investigated whether HIF inhibitors could affect several other important mediators of immune tolerance such as myeloid-derived suppressor cells (MDSC) and T regulatory cells (i.e, recruitment, function and differentiation). Overall, hypoxia inhibition in hypoxic tumors can be used as a cutting-edge approach to improve cancer immunotherapy and to formulate more effective cancer vaccine-based therapy.
Acknowledgements
This work was supported by grants from Caloust Gulbenkian Foundation, Televie (7.4517.14, 7.4606.13), Fonds National de la Recherche, Luxembourg (C12/BM/ 3962058, AFR 7842786 and 7802325) and Fondation Cancer, Luxembourg (FC/2012/02 and FC/2013/03).
References
- Hoenicke L, Zender L . Immune surveillance of senescent cells--biological significance in cancer- and non-cancer pathologies. Carcinogenesis. 2012; 33: 1123-1126.
- Corthay A . Does the immune system naturally protect against cancer? Front Immunol. 2014; 5: 197.
- Smyth MJ, Godfrey DI, Trapani JA . A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001; 2: 293-299.
- Rosenberg SA . Progress in human tumour immunology and immunotherapy. Nature. 2001; 411: 380-384.
- Watzl C, Long EO . Exposing tumor cells to killer cell attack. Nat Med. 2000; 6: 867-868.
- Di Carlo E, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P . The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001; 97: 339-345.
- Mantovani A, Cassatella MA, Costantini C, Jaillon S . Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011; 11: 519-531.
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al . The immunological synapse: a molecular machine controlling T cell activation. Science. 1999; 285: 221-227.
- Angus KL, Griffiths GM . Cell polarisation and the immunological synapse. Curr Opin Cell Biol. 2013; 25: 85-91.
- Shresta S, MacIvor DM, Heusel JW, Russell JH, Ley TJ . Natural killer and lymphokine-activated killer cells require granzyme B for the rapid induction of apoptosis in susceptible target cells. Proc Natl Acad Sci U S A. 1995; 92: 5679-5683.
- Cullen SP, Brunet M, Martin SJ . Granzymes in cancer and immunity. Cell Death Differ. 2010; 17: 616-623.
- Quail DF, Joyce JA . Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013; 19: 1423-1437.
- Palazon A, Goldrath AW2, Nizet V3, Johnson RS4 . HIF Transcription Factors, Inflammation, and Immunity. Immunity. 2014; 41: 518-528.
- Hamai A, Benlalam H, Meslin F, Hasmim M, Carre T, Akalay I, et al. Immune surveillance of human cancer: if the cytotoxic T-lymphocytes play the music, does the tumoral system call the tune? Tissue antigens. 2010; 75:1-8.
- Whiteside TL . The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008; 27: 5904-5912.
- Majmundar AJ, Wong WJ, Simon MC . Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010; 40: 294-309.
- Semenza GL . Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012; 33: 207-214.
- Baldewijns MM, van Vlodrop IJ, Vermeulen PB, Soetekouw PM, van Engeland M, de BruÏne AP . VHL and HIF signalling in renal cell carcinogenesis. J Pathol. 2010; 221: 125-138.
- Salceda S, Caro J . Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997; 272: 22642-22647.
- Patel SA, Simon MC . Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ. 2008; 15: 628-634.
- Lau KW, Tian YM, Raval RR, Ratcliffe PJ, Pugh CW . Target gene selectivity of hypoxia-inducible factor-alpha in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br J Cancer. 2007; 96: 1284-1292.
- Makino Y, Uenishi R, Okamoto K, Isoe T, Hosono O, Tanaka H, et al. Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1): a negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. The Journal of biological chemistry. 2007; 282:14073-14082.
- Noman MZ, Buart S, Van Pelt J, Richon C, Hasmim M, Leleu N, et al. The cooperative induction of hypoxia-inducible factor-1 alpha and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. Journal of immunology. 2009; 182: 3510-3521.
- Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, et al . Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011; 71: 5976-5986.
- Mathew R, White E . Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011; 21: 113-119.
- Viry E, Paggetti J, Baginska J, Mgrditchian T, Berchem G, Moussay E, et al . Autophagy: An adaptive metabolic response to stress shaping the antitumor immunity. Biochem Pharmacol. 2014; .
- Moussay E, Kaoma T, Baginska J, Muller A, Van Moer K, Nicot N, et al. The acquisition of resistance to TNFalpha in breast cancer cells is associated with constitutive activation of autophagy as revealed by a transcriptome analysis using a custom microarray. Autophagy. 2011; 7: 760-770.
- Noman MZ, Janji B, Berchem G, Mami-Chouaib F, Chouaib S . Hypoxia-induced autophagy: a new player in cancer immunotherapy? Autophagy. 2012; 8: 704-706.
- Hasmim M, Noman MZ, Lauriol J, Benlalam H, Mallavialle A, Rosselli F, et al. Hypoxia-dependent inhibition of tumor cell susceptibility to CTL-mediated lysis involves NANOG induction in target cells. Journal of immunology. 2011; 187: 4031-4039.
- Hasmim M, Noman MZ, Messai Y, Bordereaux D, Gros G, Baud V, et al . Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J Immunol. 2013; 191: 5802-5806.
- Noman MZ, Buart S, Romero P, Ketari S, Janji B, Mari B, et al. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012; 72: 4629-4641.
- Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H, et al. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res. 2008; 68: 4746-4753.
- Yamada N, Yamanegi K, Ohyama H, Hata M, Nakasho K, Futani H, et al. Hypoxia downregulates the expression of cell surface MICA without increasing soluble MICA in osteosarcoma cells in a HIF-1alpha-dependent manner. International journal of oncology. 2012; 41: 2005-2012.
- Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013; 110: 17450-17455.
- Viry E, Baginska J, Berchem G, Noman MZ, Medves S, Chouaib S, et al. Autophagic degradation of GZMB/granzyme B: a new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy. 2014; 10: 173-175.
- Baginska J, Viry E, Paggetti J, Medves S, Berchem G, Moussay E, et al. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front Immunol. 2013; 4: 490.
- Wickramarachchi DC, Theofilopoulos AN, Kono DH . Immune pathology associated with altered actin cytoskeleton regulation. Autoimmunity. 2010; 43: 64-75.
- Blanchoin L, Amann KJ, Higgs HN, Marchand JB, Kaiser DA, Pollard TD . Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 2000; 404: 1007-1011.
- Symons M, Derry JM, Karlak B, Jiang S, Lemahieu V, Mccormick F, et al. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996; 84: 723-734.
- Bouma G, Burns SO, Thrasher AJ . Wiskott-Aldrich Syndrome: Immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology. 2009; 214: 778-790.
- Bouma G, Mendoza-Naranjo A, Blundell MP, de Falco E, Parsley KL, Burns SO, et al. Cytoskeletal remodeling mediated by WASp in dendritic cells is necessary for normal immune synapse formation and T-cell priming. Blood. 2011; 118: 2492-2501.
- Notarangelo LD, Ochs HD . Wiskott-Aldrich Syndrome: a model for defective actin reorganization, cell trafficking and synapse formation. Curr Opin Immunol. 2003; 15: 585-591.
- Thrasher AJ, Burns SO . WASP: a key immunological multitasker. Nat Rev Immunol. 2010; 10: 182-192.
- Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE . Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity. 2001; 14: 315-329.
- Tskvitaria-Fuller I, Rozelle AL, Yin HL, Wülfing C . Regulation of sustained actin dynamics by the TCR and costimulation as a mechanism of receptor localization. J Immunol. 2003; 171: 2287-2295.
- Mace EM, Dongre P, Hsu HT, Sinha P, James AM, Mann SS, et al. Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol Cell Biol. 2014; 92: 245-255.
- Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM . Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006; 443: 462-465.
- Stinchcombe JC, Griffiths GM . Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol. 2007; 23: 495-517.
- Liu D, Xu L, Yang F, Li D, Gong F, Xu T. Rapid biogenesis and sensitization of secretory lysosomes in NK cells mediated by target-cell recognition. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 123-127.
- Liu D, Bryceson YT, Meckel T, Vasiliver-Shamis G, Dustin ML, Long EO . Integrin-dependent organization and bidirectional vesicular traffic at cytotoxic immune synapses. Immunity. 2009; 31: 99-109.
- Bertrand F, Müller S, Roh KH, Laurent C, Dupré L, Valitutti S . An initial and rapid step of lytic granule secretion precedes microtubule organizing center polarization at the cytotoxic T lymphocyte/target cell synapse. Proc Natl Acad Sci U S A. 2013; 110: 6073-6078.
- Billadeau DD, Nolz JC, Gomez TS . Regulation of T-cell activation by the cytoskeleton. Nat Rev Immunol. 2007; 7: 131-143.
- Burkhardt JK, Carrizosa E, Shaffer MH . The actin cytoskeleton in T cell activation. Annu Rev Immunol. 2008; 26: 233-259.
- Ritter AT, Angus KL, Griffiths GM . The role of the cytoskeleton at the immunological synapse. Immunol Rev. 2013; 256: 107-117.
- Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nature reviews Immunology. 2005; 5: 546-559.
- Orange JS, Harris KE, Andzelm MM, Valter MM, Geha RS, Strominger JL . The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proc Natl Acad Sci U S A. 2003; 100: 14151-14156.
- Orange JS . Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol. 2008; 8: 713-725.
- Sanborn KB, Rak GD, Maru SY, Demers K, Difeo A, Martignetti JA, et al. Myosin IIA associates with NK cell lytic granules to enable their interaction with F-actin and function at the immunological synapse. J Immunol. 2009; 182: 6969-6984.
- Andzelm MM, Chen X, Krzewski K, Orange JS, Strominger JL . Myosin IIA is required for cytolytic granule exocytosis in human NK cells. J Exp Med. 2007; 204: 2285-2291.
- Rak GD, Mace EM, Banerjee PP, Svitkina T, Orange JS . Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol. 2011; 9: e1001151.
- Brown AC, Oddos S, Dobbie IM, Alakoskela JM, Parton RM, Eissmann P, et al. Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy. PLoS biology. 2011; 9:e1001152.
- Mace EM, Orange JS . Dual channel STED nanoscopy of lytic granules on actin filaments in natural killer cells. Commun Integr Biol. 2012; 5: 184-186.
- Rodriguez Del Castillo A, Lemaire S, Tchakarov L, Jeyapragasan M, Doucet JP, Vitale ML, et al . Chromaffin cell scinderin, a novel calcium-dependent actin filament-severing protein. EMBO J. 1990; 9: 43-52.
- Trifaro JM, Rose SD, Marcu MG. Scinderin, a Ca2+-dependent actin filament severing protein that controls cortical actin network dynamics during secretion. Neurochemical research. 2000; 25:133-144.
- Carter N, Nakamoto T, Hirai H, Hunter T . EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas). Nat Cell Biol. 2002; 4: 565-573.
- Abouzahr S, Bismuth G, Gaudin C, Caroll O, Van Endert P, Jalil A, et al. Identification of target actin content and polymerization status as a mechanism of tumor resistance after cytolytic T lymphocyte pressure. Proc Natl Acad Sci U S A. 2006; 103: 1428-1433.
- Abouzahr-Rifai S, Hasmim M, Boukerche H, Hamelin J, Janji B, Jalil A, et al. Resistance of tumor cells to cytolytic T lymphocytes involves Rho-GTPases and focal adhesion kinase activation. J Biol Chem. 2008; 283: 31665-31672.
- Gross CC, Brzostowski JA, Liu D, Long EO . Tethering of intercellular adhesion molecule on target cells is required for LFA-1-dependent NK cell adhesion and granule polarization. J Immunol. 2010; 185: 2918-2926.
- Görgün G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG . Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest. 2005; 115: 1797-1805.
- Ramsay AG, Johnson AJ, Lee AM, Gorgün G, Le Dieu R, Blum W, et al . Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. 2008; 118: 2427-2437.
- Ramsay AG, Clear AJ, Fatah R, Gribben JG . Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012; 120: 1412-1421.
- Ramsay AG, Evans R, Kiaii S, Svensson L, Hogg N, Gribben JG . Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by altering Rho GTPase signaling that is reversible with lenalidomide. Blood. 2013; 121: 2704-2714.
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al . Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005; 8: 241-254.
- Myllyharju J, Schipani E . Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res. 2010; 339: 19-29.
- Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL . Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. 2013; 288: 10819-10829.
- Gilkes DM, Semenza GL2, Wirtz D3 . Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014; 14: 430-439.
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al . Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009; 139: 891-906.
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nature cell biology. 2013; 15: 637-646.
- Gilkes DM, Xiang L, Lee SJ, Chaturvedi P, Hubbi ME, Wirtz D, et al. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111: E384-393.
- Ridley AJ . Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006; 16: 522-529.
- Vega FM, Ridley AJ . Rho GTPases in cancer cell biology. FEBS Lett. 2008; 582: 2093-2101.
- Turcotte S, Desrosiers RR, Béliveau R . HIF-1alpha mRNA and protein upregulation involves Rho GTPase expression during hypoxia in renal cell carcinoma. J Cell Sci. 2003; 116: 2247-2260.
- Hirota K, Semenza GL . Rac1 activity is required for the activation of hypoxia-inducible factor 1. J Biol Chem. 2001; 276: 21166-21172.
- Marques CA, Hähnel PS, Wölfel C, Thaler S, Huber C, Theobald M, et al. An immune escape screen reveals Cdc42 as regulator of cancer susceptibility to lymphocyte-mediated tumor suppression. Blood. 2008; 111: 1413-1419.
- Zieseniss A. Hypoxia and the modulation of the actin cytoskeleton - emerging interrelations. Hypoxia. 2014; 2:11-21.
- Paggetti J, Viry E, Berchem G, Moussay E, Janji B. Hypoxia-induced autophagy in tumor cells: a key target for improving cancer immunotherapy. Can Cell Microenviron. 2014; 1: e213.
- Akalay I, Janji B, Hasmim M, Noman MZ, André F, De Cremoux P, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013; 73: 2418-2427.
- Akalay I, Janji B, Hasmim M, Noman MZ, Thiery JP, Mami-Chouaib F, et al. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy. 2013; 9: 1104-1106.
- Aguilera MO, Berón W, Colombo MI . The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy. 2012; 8: 1590-1603.
- Tang HW, Wang YB, Wang SL, Wu MH, Lin SY, Chen GC. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. The EMBO journal. 2011; 30: 636-651.
- Chen GC, Lee JY, Tang HW, Debnath J, Thomas SM, Settleman J . Genetic interactions between Drosophila melanogaster Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy. 2008; 4: 37-45.