
Review Article
J Immun Res. 2015;2(2):1016.
Lesson from Viral Interferon Regulatory Factors
Priyanka Sivadas and Hye-Ra Lee*
Department of Molecular Microbiology and Immunology,Keck School of Medicine, University of Southern California, Harlyne J Norris Cancer Research Tower, USA
*Corresponding author: Hye-Ra Lee, Department of Molecular Microbiology and Immunology, University of Southern California, Harlyne J Norris Cancer Research Tower, Los Angeles, USA
Received: February 01, 2015; Accepted: February 16, 2015; Published: February 17, 2015
Abstract
Once virus infects cells, the host immediately turns on their immune reponse to eliminate the virus, while the virus attempts to subvert the host immune system for survival. Therefore, kaposi’s sarcoma-associated herpesvirus (KSHV), a DNA tumor virus, dedicates a large portion of its genome to harbor immunomodulatory proteins in order to sustain efficient life long persistency as well as their life cycle. This review delineates a concise overview of the molecular events of viral interferon regulatory factors (vIRFs) underlying viral immune evasion strategies during life cycle of KSHV.
Keywords: KSHV; VIRFs; Viral immune evasion; KSHV life cycle
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV) belongs to the gamma-herpesvirus family and is known to be the causative agent of several human cancers including primary effusion lymphoma (PEL), multicentric Castleman’s disease (MCD), and kaposi’s sarcoma (KS) [1]. As with other herpesviruses, once KSHV infected the host, KSHV displays two different life cycles; latent and lytic. During the latent life cycle, it manifests persistency as well as reversibility property. Hence, viral genome exists as a circular episome in the nucleus of host cells, rigorously expressing a handful of viral genes to allow the virus to retain lifelong persistent infection [2]. Under certain circumstances, however, it can reactivate and virtually the entire set of viral genes is expressed, leading to progeny virus production. Thereby, in order to establish their efficient life cycle, KSHV harbors numerous genes that have abilities to overcome the host immune antiviral responses. One mechanism through which KSHV evades the host immune system is by encoding viral homologs of cellular genes that augment or subvert the functions of their cellular counterparts. Among them,KSHV harbors genes with significant homology to cellular interferon regulatory factors (IRFs), aptly named viral IRFs (vIRFs) [3, 4].
Remarkably, KSHV is the only human virus known to carry vIRFs and contains four different vIRFs (vIRF1-vIRF4) within a clustered locus [5, 6]. They are all expressed during lytic reactivation, but vIRF3, also called latency-associated nuclear antigen 2 (LANA2), has been detected in latently infected PEL cells [3, 4, 6]. It is indicated that they may act independently depending on the cell type and the phase of the viral life cycle. The accumulated previous studies suggested that vIRFs have developed two main strategies: First, it inhibits the IFNmediated innate immunity. Second, it represses the p53-mediated tumor suppressor activity [3]. In addition, a recent growing body of evidence is shedding light on its function as a viral transcriptional factor to regulate cellular gene expression. The goal of this review is to delineate how KSHV vIRFs modulate host immune system for their own benefits, thereby either promoting efficient viral replication or viral persistency.
Immune Responses
IFN pathway
One of the primary responses to viral infection by cells is the expression of type I IFNs (IFN-α and IFN-β) that result in the expression of genes that cause cell growth suppression, apoptosis, antigen presentation, and modulation of several signal transduction pathways [7, 8]. These genes are upregulated by IRFs, a family of transcription factors, which are activated by IFN signaling through their cognate receptor [9]. All IRFs share homology in the C-terminal region that contains the IRF-association domain (IAD) and the N-terminal region that encodes the DNA-binding domain (DBD), which is characterized by the presence of five tryptophan repeats. Among the nine members (IRF1 to IRF9) of the IRF family identified thus far, IRF3 and IFR7 are the key regulators of the expression of the IFN-α/β genes upon viral infection [7, 9]. Viral infection activates certain host pattern recognition receptors (PRRs), resulting in the phosphorylation of cytoplasmic IRF3 and its subsequent translocation to the nucleus, wherein it interacts with the transcriptional coactivator histone acetyltransferase (HAT) CBP/p300 to induce IFN-β gene expression [10, 11]. IRF7 is highly homologous to IRF3, but unlike IRF3, IRF7 is constitutively expressed at low levels in most cells and is strongly induced by the type I-IFN-mediated signaling stemming from an IRF9-dependent pathway [12-14]. Like IRF3, IRF7 undergoes phosphorylation upon viral infection, which allows its dimerization and nuclear translocation [13, 15]. IRF7 either hetero dimerizes with IRF3 or homodimerizes, with these dimers inducing the expression of chemokines and the IFN-α/β genes [13, 15]. The large set of genes then induced by IFN-α/β ultimately form the first line of the anti-viral defenses in suppressing viral replication and propagation. In turn, viruses have evolutionally developed various strategies to subvert these pathways, to the benefit of their life cycles. As an example, KSHV expresses the vIRFs to act as dominantnegative inhibitors by targeting IRF3 and IRF7 [7, 9]. Hence, it is not surprising that KSHV IRFs have evolutionarily developed various tactics to subvert these pathways, to their advantage.
vIRF1 (K9): vIRF1 has been identified as the first vIRF found to effectively repress cellular IFN responses [16, 17]. vIRF1 suppresses type I and type II IFN response and one of the known mechanisms is through inhibition of IRF1 transactivation without competing with IRF1 for DNA binding [16]. Alternatively, vIRF1 binds to transcriptional cofactor p300 and interferes with CBP/p300-IRF3 complex formation along with p300 histone acetyltransferase (HAT) activity, thus preventing IRF3-mediated transcriptional activation [18, 19]. Recently, it was shown that vIRF1, vIRF2, vIRF3 have different capability to block Toll-like receptor 3 (TLR3)-mediated IFN induction [20]. First, vIRF1 and vIRF2 inhibit transcription and translational level of IFN- upon TLR3 activation [20]. Second, only vIRF1 but not vIRF2 or vIRF3 reduced phosphorylation and nuclear translocation of IRF3 upon TLR3 activation [20]. Overall, it implies that vIRFs might possess selectivity for a specific TLR owing to inhibition of TLR-mediated IFN production.
vIRF2 (K11/K11.1): Full-length vIRF2, which is translated from exons K11.1 and K11, repressed IRF3 mediated IFN-β transcriptional activity via stimulation of IRF3 degradation and inhibition of IRF3 transactivation [21]. vIRF2 inhibits IFN-α/β driven signaling as well as signaling induced by IFN-γ [22]. The underlying mechanism, however, is yet to be defined. In addition, vIRF2 reduced the activation of the IFN-induced interferon-response element (ISRE) promoter through the deregulation of IFN-stimulated gene factor-3 (ISGF-3) [23]. It is suggested that vIRF2 possesses pleiotropic activity of inhibiting early type I IFN (IFN enhancesome-dependent) and delayed type I IFN (ISGF-dependent) responses. Furthermore, previous studies have shown that the first exon of vIRF2 (K11.1) prevents dsRNA-activated protein kinase (PKR) kinase activity [24], reducing protein synthesis and blocking IFN-α/β signaling to decrease the ability of cells to respond to viral infections [25, 26]. In binding assay, this short form of K11.1 interacts with cellular IRF1, IRF2, IRF8, RelA, and p300, but not IRF3.
vIRF3 (K10.5): vIRF3 interaction with cellular IRF7, suppresses IRF7 DNA binding activity and, therefore, inhibits IFN-mediated immunity through the inhibition of IFN-α production [27]. Remarkably, a putative double α-helix motif of vIRF3 (residues 240- 280) that has been shown to be responsible for the interaction of vIRF3 with IRF7 is also sufficient to bind to IRF5 [28]. As a result of this interaction, vIRF3 inhibits IRF5-mediatd ISRE and IFN-β promoter activity [28]. It was recently shown that vIRF3 is required for the survival of PEL cells. RNA interference (RNAi) knockdown of vIRF3 in PEL cells reduced cell proliferation by releasing IRF5 from p21 promoter transcription complexes. Moreover, vIRF3 inhibits the function of the IFN-induced PKR. PKR inhibits viral mRNA translation by phosphorylating eIF-2 and regulates host defense by controlling transcription [29].
In summary, the downregulation of the IFN regulatory pathway is a common characteristic of the three vIRFs whose functions have been well studied, as IRF3 and IRF7 are the initial key factors of the host immune surveillance program against viral infections (Table 1). While it remains to be discovered whether vIRF4, the most recently identified member of the vIRF family, affects IFN-mediated innate immunity, the down-regulation of the IFN regulatory pathway is a common characteristic of vIRFs, as IRF3 and IRF7 are key initiation factors of the host immune surveillance program against viral infection.
![]()
Anti-immune strategies of KSHV vIRFs
Strategy
Gene product
Function
Innate
Inhibit interferon production
vIRF1
Inhibition of IRF3-mediated transcription
vIRF2
Suppression of IRF3
Degradation of ISGF-3
Prevent PKR activity
vIRF3
Inhibition of IRF7 DNA binding activity
Regulation of p53 tumor suppression
vIRFs
Suppression of p53
Regulation of CD95/CD95L pathway
vIRF1
Inhibition of CD95L induction
vIRF2
Regulation of apoptosis pathway
vIRF1
Inhibition of Bim-mediated cellular pro-apoptotic function
Inhibition of GRIM-19-mediated cellular apoptotic function
Deregulation of gene expression
vIRF4
Downregulation of c-Myc and IRF4
Adaptive
Inhibit MHC-I and MCH-II
vIRF1
Downregulates MHC class I
vIRF3
Downregulates MHC class II
Table 1: Summary of immune evasion function by KSHV vIRFs.
Furthermore, KSHV vIRFs also employ another way to efficiently regulate Type I IFN signaling. Accumulated data shows that expression of KSHV vIRF1, -2, or -3 blocks TLR3-mediated activation of IFN-responsive promoter activity. Remarkably, both vIRF1 and vIRF2 inhibit IFN-b production upon TLR3 activation (20). However, it seems that vIRF1 and vIRF2 block TLR3-mediating signaling via different mechanisms: expressing vIRF1 but not vIRF2 led to decreased phosphorylation and nuclear translocation of IRF3 in response to TLR3 activation (20). It is indicated that vIRFs also employed capability to block TLR3-mediated IFN pathway.
MHC down regulation
The major histocompatibility complex, MHC-I and MHCII molecules control a major part of the host immune response to pathogen by virtue of their ability to present peptides to CD8+ and CD4+ T cells, respectively. Hence, KSHV escapes from MHC-I and MHC-II peptide presentation to avoid recognition and increase virus survival. Notably, vIRFs are one of KSHV proteins that can also regulate host adaptive immune response through deregulation of MHC-I and MHC–II expression.
vIRF1 (K9): vIRF1 has capacity to downregulate either IFNor NF-κB-mediated induction of MHC-I expression through its interaction with transcriptional co activator p300 in lymphatic endothelial cells (LECs) [30]. In contrast, the KSHV latent protein, vFLIP, upregulates MHC-I by NF-κB activation [30]. It suggests that KSHV employs vIRF1 to orchestrate balance between immune evasion and immune activation leading to optimal coexistence with its host.
vIRF3 (K10.5): The MHC-II gene encodes the polymorphic HLADR, -DQ, and -DP molecules, which are expressed as α- and β-chain heterodimers on the cell surface. In contrast to the classical MHC-I molecules, which are constitutively expressed on almost all cells, the constitutive expression of MHC-II molecules is tissue-specific and is restricted to professional antigen presenting cells and in thymic epithelial cells [31]. However, all other cell types lack constitutive expression of MHC-II molecules, but their expression can be induced by exposure to cytokines such as IFN-γ, a well known transcriptional factor of MHC-II [31]. Thereby, vIRF3 reduces the transcriptional activity of IFN-γ, resulting in downregulation of MHC-II expression [32]. Recently, Zuo et. al showed that expression of vIRF3 suppresses surface HLA-DR, and subsequently impedes recognition by KSHVspecific CD4+ T cells [33]. Given critical role of MHC-I and -II, these genes are tightly regulated at the transcriptional level by a variety of transcription factors. For instance, in humans, MHC-II expression is inducible by IFN-γ in almost every cell type [31]. vIRF3 inhibits the expression of MHC-II due to reduced activity in IFN-γ, which was verified by both IFN-γ promoter assays and by analyzing the expression of common target genes upon knockdown of vIRF3 (32). In addition, vIRF3-mediated inhibition of CIITA transcription, known master regulator of MHC-II, substantially downregulates MHC-II expression [32, 33]. Taken together, vIRF3 utilizes two different strategies to effectively suppress MHC-II expression via IFN-γ as well as CIITA. This suggests that vIRF3 might function as a critical determinant of MHC-II antigen presentation that is likely important to escape the immune surveillance on maintenance of latency and survival of infected cells.
Cell Death Pathway
Regulation of p53 tumor suppression
Exposure to cellular responses can trigger p53, a transcriptional factor, to induce cell cycle arrest, cellular senescence, or apoptosis to protect cells from many different types of stress induced damages, and among these viral infections represents a major type of cellular stress [34]. Hence, in order to counteract host innate immune response, viruses openly employ genes that inhibit p53-mediated growth suppressive and proapoptotic actions [2, 35, 36].
Under normal conditions, p53 has a short half-life and its intracellular basal levels are very low [34, 37-39]. However, stressful conditions, such as a viral infection, leads to stabilization of p53 protein, resulting in activation of p53, and ultimately promption of cell cycle arrest or apoptosis [34, 39]. Notably, theses changes in p53 are mediated by extensive post-translational modification of p53 and these protein modifications appear to lead the activation of p53 protein in two ways: first the half-life of p53 increases, leading to increased level of p53 protein. Second, p53 binds to specific DNA sequences and promotes the transcription of downstream genes, such as p21, MDM2, BAX, and so on [34, 39]. Thus in order to hinder p53- mediated irreversible cell cycle arrest and apoptosis, viruses need to tightly regulate the expression and/or function of p53, which KSHV accomplishes through its vIRFs [2, 3, 36].
vIRF1 (K9): vIRF1 inhibits activation of p53 through two engaged mechanisms. First, vIRF1 interacts with DNA-binding domain (DBD) of p53, thereby suppressing its acetylation and this interaction inhibits p53-mediated transcriptional activation of its target genes, for instance p21 and BAX [40, 41]. Second, later reports indicated that vIRF1 interacts with ATM kinase and blocks ATM activation, which resulted in the reduction of Ser15 phosphorylation and subsequently enhanced degradation of p53 [42]. Collectively, vIRF1 comprehensively inhibits p53 tumor suppressor function by downregulation of its transcriptional activity as well as its protein stability.
vIRF3 (K10.5): vIRF3 was shown to interact with p53 and inhibit p53-mediated apoptosis [43]. Additionally, this interaction decreases the activation of caspase-8 by p53 [43]. However, the molecular mechanism underlying this phenomenon is not known. Very recently, Baresova et. al revealed how vIRF3 targets p53 pathway [4]. vIRF3 binds to the DBD of p53, impairs p53 oligomerization and DNA-binding ability, resulting in decreased transcriptional activation of p53 target genes including growth-regulatory p21 gene. Furthermore, vIRF3 inhibits ATM autophosphorylation and Ser15/Ser20 phosphorylation of p53, which consequently leads to an increase in p53 ubiquitination [4]. More importantly, the knockdown of vIRF3 using RNA interference (RNAi) in KSHV positive PEL cells restores the p53 protein levels, p53 phosphorylation, and ATM autophosphorylation, accompanied by an increase in p21 gene expression [4]. Hence, like vIRF1, vIRF3 employs a very efficient way to downregulate p53 activity and thus promotes cell proliferation to maintain KSHV persistency.
vIRF4 (K10): vIRF4 interacts with murine double minute 2 (MDM2), known E3 ligase of p53, dramatically increases stability of MDM2 through blocking its autoubiquitination [44]. Consequently, vIRF4 expression markedly enhances p53 ubiquitination and degradation, effectively suppressing p53-mediated apoptosis [44]. Furthermore, vIRF4 interacts with herpesvirus-associated ubiquitinspecific protein (HAUSP) that is capable of dual control of both p53 and its negative regulator MDM2 [45]. Unlike vIRF4 mutant which no longer interacts with MDM2, vIRF4 mutant, which no longer interacts with HAUSP, minorly increase p53 ubiquitination [45]. Thus, although vIRF4 interacts with both HAUSP and MDM2 to control p53 levels, this report indicates that the vIRF4-MDM2 interaction is more important in p53 downregulation than vIRF4- HAUSP interaction [45]. Taken together, KSHV vIRF4 is a strong anti-apoptotic factor, thereby it comprehensively blocks p53 activity in a genetically separable and functionally additive manner: N-terminal region of vIRF4 keeps HAUSP in check, while the C-terminal region of vIRF4 inhibits MDM2.
Regulation of CD95/CD95L signaling pathway
Apoptosis is triggered by two major apoptosis initiating pathways, designated as intrinsic and extrinsic pathway. Especially, the extrinsic pathway is activated by the binding of apoptotic ligands to death receptors, such as TNF receptor superfamily, on the cell surface [46]. CD95 (APO-1/Fas), a type I transmembrane protein, is a member of the TNF receptor superfamily expressed in various tissues, while expression of its cognate ligand (CD95L), a type II transmembrane protein, is tightly regulated by multiple transcription factors including NF-κB, forkhead transcription factors, and IRF1 [47-50]. CD95L induces apoptosis via formation of a death-inducing signaling complex and initiation of a signaling cascade of caspases [51, 52].
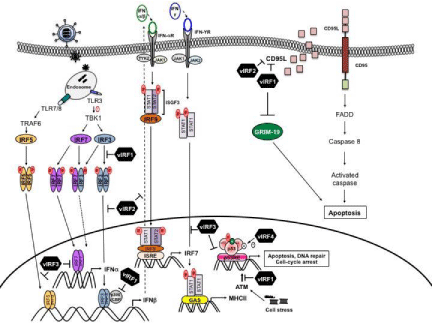
Figure 1: Schematic representation of KSHV immune evasion strategies.
vIRF1 (K9) and vIRF2 (K11/K11.1): vIRF1 and vIRF2 inhibit IRF-1-mediated and TCR/CD3-mediated induction of the CD95L, which resulted in strong reduction of activation-induced cell death [48]. vIRF1 prevents CD95L expression by inhibition of binding of IRF-1 with positive regulatory IRF-1-dependent domains (PRIDDs) in CD95L promoter, however the mechanism is not clear. One potential mechanism is that vIRF2 inhibits NF-κB, known transcriptional factor of CD95L.
The other cell death pathway
vIRF1 was also shown to associate with the pro-apoptotic BH3- only Bcl-2 family member, Bim, which leads to localization of Bim to the nucleus [53]. Thereby, it inhibits Bim-mediated cellular proapoptosis signal and consequently increases viral production [53]. Moreover, to understand function of vIRF1, Seo et al. performed a yeast-two hybrid screen and identified the interferon/retinoid acid (IFN/RA)-inducible cell death regulator (GRIM)-19 as novel binding partners of vIRF1 [54]. GRIM-19 was isolated as a death-associated gene that enhanced apoptotic cell death in response to IFN-β and retinoic acid (RA) [55]. In the context of its immunomodulatory function, vIRF1 directly binds to GRIM-19 and blocks its ability to induce apoptosis [54].
Effects on Gene Expression
As described earlier, IRFs are well known transcription factors that play an important role in the induction of genes encoded by IFNs. Each IRF contains well-conserved transactivation domains at C-terminal region and DNA binding domain with five tryptophan repeats, which form a helix-turn-helix motif and bind similar DNA sequence, designated as IFN-stimulated response element [56]. Unlike cellular IRFs, vIRFs only carry three tryptophan residues, indicating that it has potency to bind DNA. However, it is not clear whether vIRFs directly interact with DNA sequence similar to their cellular counter part, IRFs [35]. Recently, our group has shown that vIRF4 grossly reshapes host gene expression, ultimately leading to favorable circumstance for viral life cycle [57]. It is interesting to note that, vIRF4 not only reduces cellular IRF4 (c-IRF4) expression but also with c-IRF4 for binding to specific promoter region of the c-Myc gene, resulting in drastic suppression of c-Myc expression [57]. vIRF4 vigorously downregulates c-Myc and c-IRF4 expression, which ultimately facilitates KSHV lytic replication [57]. Especially, vIRF4 putative DNA binding domain region is necessary to deregulate both c-Myc and c-IRF4. Consequently, this study collectively suggests the potential important role of vIRFs as a viral transcriptional factor. Moreover, it appears that vIRF1 binds to a specific consensus sequence of viral DNA [58]. Thus, collectively, it indicates that vIRFs might be involved in transcriptional regulation and expression of both cellular and viral genes and function as a viral transcriptional factor.
Conclusion
In this review, we have briefly described common functions of KSHV vIRFs, inhibition of IFN-mediated immunity and p53- mediated tumor suppressor function (Table 1). Most notably, a recent growing body of information indicates that each vIRF delineates a unique ability to deregulate specific cellular function with respect to innate and adaptive immune system. Furthermore, vIRFs have been shown to have the capability to bind promoter regions of both cellular and viral genes to meticulously reshape the cellular environment by reprogramming host gene expression, while synergistically regulating viral protein transcription [57, 59], thereby either promoting efficient viral replication or facilitating viral persistence. Further work is necessary to determine vIRFs function as a viral transcriptional factor and it will ultimately pave the way to understand why KSHV encodes multiple copies of vIRFs and their impact on KSHV viral life cycle and pathogenesis. Thus, understanding the biological foundation of vIRFs will yield further information on novel strategies used by KSHV to modulate host immunity and influence virus-induced pathology.
References
- Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest. 2010; 120: 939-949.
- Lee HR, Brulois K, Wong L, Jung JU. Modulation of Immune System by Kaposi's Sarcoma-Associated Herpesvirus: Lessons from Viral Evasion Strategies. Front Microbiol. 2012; 3: 44.
- Lee HR, Kim MH, Lee JS, Liang C, Jung JU. Viral interferon regulatory factors. J Interferon Cytokine Res. 2009; 29: 621-627.
- Baresova P, Musilova J, Pitha PM, Lubyova B. p53 tumor suppressor protein stability and transcriptional activity are targeted by Kaposi's sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3. Mol Cell Biol. 2014; 34: 386-399.
- Takaoka A, Tamura T, Taniguchi T. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. 2008; 99: 467-478.
- Cunningham C, Barnard S, Blackbourn DJ, Davison AJ. Transcription mapping of human herpesvirus 8 genes encoding viral interferon regulatory factors. J Gen Virol. 2003; 84: 1471-1483.
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000; 13: 539-548.
- McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015; 15: 87-103.
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005; 434: 772-777.
- Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol. 1998; 18: 2986-2996.
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998; 17: 1087-1095.
- Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998; 441: 106-110.
- Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998; 17: 6660-6669.
- Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006; 6: 644-658.
- Lin R, Mamane Y, Hiscott J. Multiple regulatory domains control IRF-7 activity in response to virus infection. J Biol Chem. 2000; 275: 34320-34327.
- Zimring JC, Goodbourn S, Offermann MK. Human herpesvirus 8 encodes an interferon regulatory factor (IRF) homolog that represses IRF-1-mediated transcription. J Virol. 1998; 72: 701-707.
- S. J. Gao et al., Oncogene 15, 1979 (Oct 16, 1997).
- Li M, Damania B, Alvarez X, Ogryzko V, Ozato K, Jung JU. Inhibition of p300 histone acetyltransferase by viral interferon regulatory factor. Mol Cell Biol. 2000; 20: 8254-8263.
- Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ Jr, et al. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene. 2001; 20: 800-811.
- Jacobs SR, Gregory SM, West JA, Wollish AC, Bennett CL, Blackbourn DJ, et al. The viral interferon regulatory factors of kaposi's sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J Virol. 2013; 87: 798-806.
- Aresté C, Mutocheluh M, Blackbourn DJ. Identification of caspase-mediated decay of interferon regulatory factor-3, exploited by a Kaposi sarcoma-associated herpesvirus immunoregulatory protein. J Biol Chem. 2009; 284: 23272-23285.
- Fuld S, Cunningham C, Klucher K, Davison AJ, Blackbourn DJ. Inhibition of interferon signaling by the Kaposi's sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J Virol. 2006; 80: 3092-3097.
- Mutocheluh M, Hindle L, Aresté C, Chanas SA, Butler LM, Lowry K, et al. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor-2 inhibits type 1 interferon signalling by targeting interferon-stimulated gene factor-3. J Gen Virol. 2011; 92: 2394-2398.
- Burýsek L, Pitha PM. Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J Virol. 2001; 75: 2345-2352.
- Burýsek L, Yeow WS, Lubyová B, Kellum M, Schafer SL, Huang YQ, et al. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J Virol. 1999; 73: 7334-7342.
- L. Burysek, W. S. Yeow, P. M. Pitha, J Hum Virol 2, 19 (Jan-Feb, 1999).
- Joo CH, Shin YC, Gack M, Wu L, Levy D, Jung JU. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's sarcoma-associated herpesvirus viral IRF homolog vIRF3. J Virol. 2007; 81: 8282-8292.
- Wies E, Hahn AS, Schmidt K, Viebahn C, Rohland N, Lux A, et al. The Kaposi's Sarcoma-associated Herpesvirus-encoded vIRF-3 Inhibits Cellular IRF-5. J Biol Chem. 2009; 284: 8525-8538.
- Esteban M, García MA, Domingo-Gil E, Arroyo J, Nombela C, Rivas C. The latency protein LANA2 from Kaposi's sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J Gen Virol. 2003; 84: 1463-1470.
- Lagos D, Trotter MW, Vart RJ, Wang HW, Matthews NC, Hansen A, et al. Kaposi sarcoma herpesvirus-encoded vFLIP and vIRF1 regulate antigen presentation in lymphatic endothelial cells. Blood. 2007; 109: 1550-1558.
- P. J. van den Elsen, Front Immunol 2, 48 (2011).
- Schmidt K, Wies E, Neipel F. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 3 inhibits gamma interferon and major histocompatibility complex class II expression. J Virol. 2011; 85: 4530-4537.
- Zuo J, Hislop AD, Leung CS, Sabbah S, Rowe M. Kaposi's sarcoma-associated herpesvirus-encoded viral IRF3 modulates major histocompatibility complex class II (MHC-II) antigen presentation through MHC-II transactivator-dependent and -independent mechanisms: implications for oncogenesis. J Virol. 2013; 87: 5340-5350.
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000; 408: 307-310.
- Lee HR, Lee S, Chaudhary PM, Gill P, Jung JU. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Future Microbiol. 2010; 5: 1349-1365.
- Coscoy L. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Nat Rev Immunol. 2007; 7: 391-401.
- Brooks CL, Gu W. p53 regulation by ubiquitin. FEBS Lett. 2011; 585: 2803-2809.
- Collot-Teixeira S, Bass J, Denis F, Ranger-Rogez S. Human tumor suppressor p53 and DNA viruses. Rev Med Virol. 2004; 14: 301-319.
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005; 24: 2899-2908.
- Seo T, Park J, Lee D, Hwang SG, Choe J. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J Virol. 2001; 75: 6193-6198.
- Nakamura H, Li M, Zarycki J, Jung JU. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J Virol. 2001; 75: 7572-7582.
- Shin YC, Nakamura H, Liang X, Feng P, Chang H, Kowalik TF, et al. Inhibition of the ATM/p53 signal transduction pathway by Kaposi's sarcoma-associated herpesvirus interferon regulatory factor 1. J Virol. 2006; 80: 2257-2266.
- Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J Virol. 2001; 75: 429-438.
- Lee HR, Toth Z, Shin YC, Lee JS, Chang H, Gu W, et al. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J Virol. 2009; 83: 6739-6747.
- Lee HR, Choi WC, Lee S, Hwang J, Hwang E, Guchhait K, et al. Bilateral inhibition of HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat Struct Mol Biol. 2011; 18: 1336-1344.
- Fouqué A, Debure L, Legembre P. The CD95/CD95L signaling pathway: a role in carcinogenesis. Biochim Biophys Acta. 2014; 1846: 130-141.
- Kirchhoff S, Sebens T, Baumann S, Krueger A, Zawatzky R, Li-Weber M, et al. Viral IFN-regulatory factors inhibit activation-induced cell death via two positive regulatory IFN-regulatory factor 1-dependent domains in the CD95 ligand promoter. J Immunol. 2002; 168: 1226-1234.
- Kasibhatla S, Genestier L, Green DR. Regulation of fas-ligand expression during activation-induced cell death in T lymphocytes via nuclear factor kappaB. J Biol Chem. 1999; 274: 987-992.
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999; 96: 857-868.
- Chow WA, Fang JJ, Yee JK. The IFN regulatory factor family participates in regulation of Fas ligand gene expression in T cells. J Immunol. 2000; 164: 3512-3518.
- Krammer PH. CD95(APO-1/Fas)-mediated apoptosis: live and let die. Adv Immunol. 1999; 71: 163-210.
- Krammer PH, Galle PR, Möller P, Debatin KM. CD95(APO-1/Fas)-mediated apoptosis in normal and malignant liver, colon, and hematopoietic cells. Adv Cancer Res. 1998; 75: 251-273.
- Choi YB, Nicholas J. Bim nuclear translocation and inactivation by viral interferon regulatory factor. PLoS Pathog. 2010; 6: e1001031.
- Seo T, Lee D, Shim YS, Angell JE, Chidambaram NV, Kalvakolanu DV, et al. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus interacts with a cell death regulator, GRIM19, and inhibits interferon/retinoic acid-induced cell death. J Virol. 2002; 76: 8797-8807.
- Moreira S, Correia M, Soares P, Máximo V. GRIM-19 function in cancer development. Mitochondrion. 2011; 11: 693-699.
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014; 32: 513-545.
- Lee HR, DoÄŸanay S, Chung B, Toth Z, Brulois K, Lee S, et al. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4) targets expression of cellular IRF4 and the Myc gene to facilitate lytic replication. J Virol. 2014; 88: 2183-2194.
- Park J, Lee MS, Yoo SM, Jeong KW, Lee D, Choe J, et al. Identification of the DNA sequence interacting with Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 1. J Virol. 2007; 81: 12680-12684.
- Xi X, Persson LM, O'Brien MW, Mohr I, Wilson AC. Cooperation between viral interferon regulatory factor 4 and RTA to activate a subset of Kaposi's sarcoma-associated herpesvirus lytic promoters. J Virol. 2012; 86: 1021-1033.