
Special Article - Inflammation
J Immun Res. 2015; 2(2): 1022.
Inflammation in Alzheimer’s Disease: Cross-talk between Lipids and Innate Immune Cells of the Brain
Loewendorf AI, Fonteh AN, Harrington MG and Csete ME*
Huntington Medical Research Institutes, California, USA
*Corresponding author: Csete ME, Huntington Medical Research Institutes, California, USA
Received: July 08, 2015; Accepted: September 11, 2015; Published: September 12, 2015
Abstract
Alzheimer’s disease (AD) is distressingly common and age is the major risk factor. One of the challenges in AD research is the scarcity of information on the healthy aging brain, since many of features considered part of ‘AD pathology’ – inflammation and oxidant stress – are also present in cognitively normal elderly populations. For this reason it is critical to study AD (and pre-AD) subjects in the context of age-matched controls, an essential feature of the data set which inspired this review. Our study of lipids in cerebrospinal fluid (CSF) of aged subjects is a novel data set, especially as lipids are relatively understudied in AD, with much of the experimental and clinical research literature focused on A-beta and tau. Inflammation too is often discussed as a consequence of rather than active participant in AD pathology. For these reasons we focus on the interplay between inflammation and lipids in the pathological process of AD, an interaction that is likely important in pathophysiology of AD. We present a summary of the complex CSF lipid changes found in our clinical AD studies, and focus in on a few of these changes to highlight the importance of lipid interactions with the brain immune system in the pathogenesis of AD, recognizing that our interpretation of these data requires further study. Neither the full complexity of the brain immune system nor the changes in lipids in CSF can be reviewed here, but we hope that the many interactions highlighted between lipids and the immune system will prompt others to investigate these pathophysiologic connections, leading to a greater understanding of the causes of AD.
Keywords: Inflammation; Alzheimer’s disease; Innate immune cells; Lipids; Brain
Introduction
The immune/inflammatory system functions optimally when it is short-lived transiently activated in response to toxic or infectious stimuli. When chronically turned on, inflammation is pathologic. For example, the unchecked T cell and autoimmune response to lipid components of myelin is a major driver of multiple sclerosis [1]. Increasingly, chronic inflammation is recognized as part and parcel of neurodegenerative diseases including AD [2]. Immune responses target lipids (though protein targets dominate the literature). Lipids also initiate immune responses against other molecules in AD. Lipids can be both pro- or anti-inflammatory in the setting of the aged brain. Some lipid inflammatory mediators are likely present because they represent a ‘normal’ response to pathologic proteins that accumulate, but other lipid inflammatory mediators also drive the disease. Importantly, lipid per oxidation reactions is key triggers of neurodegeneration in AD [3] as well as drivers of atherosclerosis [4]. Post-mitotic neurons cannot regenerate and so are chronically subject to local oxidative and inflammatory injury.
Lipids constitute about half the dry mass of the brain, and in the plasma membrane they set the scene for antigen contact by resident immune cells of the brain. The activity of proteolytic enzymes, receptor (transmembrane) organization and function, and initiation of intracellular signaling pathways are all significantly influenced by plasma membrane lipid composition. Accumulation of plaque is not only generically toxic but leads to specific lipid dysfunction by altering the bulk properties of the exquisitely organized plasma membrane including its fluidity [5].
Though inflammatory lipids have been identified in brains of AD patients, their potential immunomodulatory properties have not been reviewed in detail. We recently characterized myriad changes in lipids in the cerebrospinal fluid (CSF) of AD subjects, changes that suggest a major role for inflammatory lipids as drivers of AD pathology (See Table 1 and Figures 1-3). Our studies reveal significant lipid changes in CSF in pre-clinical AD subjects, including glycerophospholipids, sphingolipids, sphingomyelinase (generally reduced in AD) and phospholipase A2 (significantly increased in AD). For this reason we focus here on lipid mediators of inflammation, particularly drilling down into lipid pathways that we have found in the CSF of subjects with pre-clinical AD but not in the CSF of elderly subjects with healthy brains [6-8].
![]()
Lipids in CSF fractions
MCI ( n=40)
versus
CN (n=70)
LOAD (n= 29) versus
CN (n=70)
LOAD (n=29)
versus
MCI (n=40)
Supernatant fraction
Phosphatidylcholine (PC
Phosphatidylethanolamine (PE)
Phosphatidylserine (PS)
Lysophosphatidylcholine (LPC)
LPC/PC
Sphingomyelin (SM)
Ceramide (CM)
Dihydroceramide (dhCM)
SM/CM
Alpha linolenic acid (C18:3n-3)
Docosahexaenoic acid (C22:6n-3)
Particle fraction
LPC/PC
Unknown PE peak
Ceramide
Pentadecanoic acid (C15:0)
Pentadecenoic acid (C15:1)
Hexadecenoic acid (palmitoleic acid, C16:1)
Heptadecanoic acid (margaric acid, C17:0)
Nanodecenoic acid (C19:1)
Eicosadienoic acid (C20:2n-6)
Eicosatrienoic acid (C20:3n-3)
Docosatetraenoic acid (C22:4n-6)
Docosapentaenoic acid (C22:5n-3)
Free fatty acids
Decanoic acid (capric acid, C10:0)
Undecanoic acid (C11:0)
Hexadecanoic acid (palmitic acid C16:0)
Hexadecenoic acid (C16:1)
Octadecanoic acid (stearic acid, C18:0
Eicosadienoic acid (C20:2n-6)
Docosahexaenoic acid (C22:6n-3)
Enzyme activities
Phospholipase A2 activity (PLA2)
*
*
Acid Sphingomyelinase activity (aSMase)
*
*
Lipid metabolism is significantly altered in CSF of AD subjects. We studied glycerophospholipids (GP), sphingolipids, and fatty acids in CSF fractions from cognitively normal (CN), mild cognitive impairment (MCI), or study participants diagnosed with late onset Alzheimer’s disease (LOAD) [9]. Several GP classes [phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS)] decreased progressively in cerebrospinal fluid (CSF) from participants with LOAD and MCI compared with CN [6]. Most PC species decreased in similar patterns but plasmalogen accounted for the greatest decrease in PE. Changes in GPs was accompanied by a significant increase in phospholipase A2 activity [6], implying upregulation of pro-inflammatory arachadonic acid.
Sphingolipid (SP) composition of CSF-derived particles [12] and supernatant differed in subjects with cognitive impairment. Acid sphingomyelinase (aSMase) but not neutral sphingomyelinase activity was lower in LOAD compared with CN and MCI. aSMase activity also correlated with amyloid beta42 (Aβ42) levels in CSF from CN subjects but not from MCI or AD study participants [8].
Odd-number chain saturated fatty acids, monounsaturated fatty acids, and polyunsaturated fatty acids (PUFA) were also altered in CSF fractions of cognitively impaired subjects [7]. Depletion of docosahexaenoic acid (DHA, 22:6n-3) shows the importance of omega-3 fatty acids in cognitive function and suggests that disturbed PUFA metabolism partly explains global AD membrane dysfunction.
Table 1: Lipids that significantly change in CSF fractions from cognitively normal (CN), mild cognitive impairment (MCI), and late onset Alzheimer’s disease (LOAD).
For reference, Table 1 is a summary of CSF lipid profiles from elderly cognitively normal (CN) compared with lipid profiles of elderly subjects with mild cognitive impairment (MCI), those with pre-clinical late onset AD (LOAD), and patients with symptomatic LOAD and memory loss [9]. This table organizes results of several of our published studies, where more detail can be found [6-8,10- 12]. We highlight some of these changes as particularly important in inflammation here. In addition Figures 1-4 contain more detail than can be discussed in the text, as a point of reference for readers interested in particular inflammatory lipids.
The Peripheral Immune System and Brain Immune Privilege
The brain has been considered ‘immune privileged’ as the bloodbrain barrier (BBB) was thought to exclude classical T cell cytotoxic responses. However, even a healthy BBB allows some peripheral immune cells into the brain [13]. In murine models, mechanisms of immune cell ‘selectivity’ of the BBB have been deciphered to some degree. For example, entry of antigen-specific CD8+ T cells into the brain depends on expression of major histocompatibility complex (MHC) class I on the luminal surface of brain endothelium [14,15]. The anatomic sites of leukocyte entry into CNS were thought to be choroid plexus and leptomeningeal vessels, or the perivascular space [16]. These sites of brain entry and exit for leukocytes were deemed necessary because of the absence of a brain lymphatic system. Recent demonstration of functional brainlymphatics lining the dural sinuses of mice raises questions about just how segregated the brain immune system is from its peripheral counterpart [17], and the hunt is certainly on for a human brain lymphatic homolog. Another recent intriguing finding, that of regulatory T cells in rat brain [18] suggests a more open BBB than that currently envisioned and highlights the continuous interaction of the peripheral immune system and CNS. Activated peripheral T cells express molecules such as integrins that enhance ability of these cells to cross the BBB into the brain, and systemic mediators can increase the general leakiness of the BBB, as occurs with sepsis. [Sepsis is associated with encephalopathy, and cognition is often permanently impaired after sepsis [19]. The peripheral immune system may well be involved in this process. Sepsis models are often used to study cognitive dysfunction, but we do not focus on them here, in favor of more direct AD models. Importantly, the BBB also limits toxin entry into the brain, perhaps its original function, which in turn limits local inflammatory responses in the brain [20].
Declining integrity of the BBB with age has been suspected for decades though little detail was available about the nature of this decline and the changes were generally considered subtle. Nonetheless, with aging as the main risk factor for AD combined with well-known age-related endothelial dysfunction, BBB dysfunction has often been invoked as a potential contributor to AD. Recently some important details of age-related BBB dysfunction have emerged: The first site of functional BBB decline in normal elderly subjects (no cognitive impairment) is the hippocampus, which is also affected early in AD. Onset of symptomatic AD is associated with further worsening of BBB integrity in hippocampus [21]. BBB disruption in turn can activate the peripheral immune system in response to systemic release of S100B into the circulation, driving a feed-forward activation of inflammation [22] that gets turned back on the brain.
The two-hit hypothesis [23] proposes that damage to the blood brain barrier (capillary endothelial cells, pericytes, and basement membrane) is an early process [21] that causes dysfunction of the neurovascular unit (vascular cells, glial cells, and neurons). Loss of the BBB integrity allows toxins, pathogens, and cells to enter the brain and interfere with neurons and brain circuit physiology. Since lipids play a major role in the health of the BBB, the lipid changes that we report in CSF and nanoparticles (Table 1 & Figure 3) may stimulate research into their potential roles in vascular pathology of AD, as discussed below.
The Role of Inflammatory Lipids in the Regulation of Cerebral Blood Flow
Cerebral circulation is tightly organized: the distance between neuron somata and the nearest micro vessel is no greater than 15 nm to facilitate O2 and nutrient availability in the high-O2 utilizing brain [24,25]. In the peripheral circulation, temporary closing of capillaries or recruitment of new ones matches O2 availability to demand. In the brain, the red blood cell (RBC) transit time varies between capillaries and the O2 demand is likely matched by regulating these transit times [26]. Although the brain receives 15% of cardiac output, cerebrovascular resistance in healthy brain is low. Vasodilation can be induced by NO from endothelial cells, adenosine and lactate end-products of metabolic activity, and vasoactive signals delivered by the neurovascular unit (NVU, composed of endothelial cells, pericytes, neurons and astrocyte end-feet) [27,28]. Signal delivery by the NVU begins with a Ca++ rise in the synaptic end-feet of astrocytes in response to synaptic activity. Propagated Ca++ activates phospholipase A2 triggering the generation of vasoactive arachadonic acid (AA) metabolites in the astrocyte end-feet surrounding arterioles and capillaries (via cyclooxygenase and epoxygenase) [29-30]. Additionally, AA diffusing to adjacent vascular smooth muscle cells or pericytes is metabolized to 20-HETE by ω-hydroxylase [28,31]. Vascular dilation or constriction occurs depending on whether PGE2/ EET or 20-HETE synthesis dominates. The repertoire of chemicals used in control of blood flow in the brain shows regional differences, for example the NO pathway is more dominant in the cerebellum while COX-2 is more active in the sensory cortex [32].
Cerebrovascular pathophysiology in late onset Alzheimer’s disease is multi factorial, with genetic predisposition (ApoE-e4), systemic risk factors (including elevated blood pressure, blood glucose and lipids), and cerebral amyloid angiopathy (aggregated proteins that include Aβ in the vessel walls) [33]. Manifestations of this vascular contribution are reduced cerebral blood flow, ischemia, damaged BBB tight junctions, focal inflammation, infarction, and increased hemorrhage [34]. The protective effect of physical exercise on AD seen in many studies may be due in part to the anti-inflammatory effects of exercise [35]. Epoxyeicosatrienoic acids (EETs) are synthesized in vascular endothelial cells and, with cholinergic (muscarinic) stimulation, induce vasorelaxation [36,37]. Co-cultures of astrocytes and endothelial cells showed that exogenous EETs and EETs from astrocytes promote mitosis and angiogenesis of cerebral endothelial cells [38]. EETs are regulated by brain endothelial specific cytochrome P450 enzymes [39].
It remains unclear if vascular pathology is a primary mechanism leading to AD or secondary to the amyloid β and tau accumulation. Clinical data demonstrate that vascular pathology and vascular risk factors are both associated with dementia, and both increase with age [40]. Recent studies are teasing out the relatedness and independence of vascular and neuronal mechanisms. For instance, brain amyloid deposition in vivo (Pittsburgh compound B) neither predicted cognitive impairment, nor did it interact with vascular brain injury (infarcts by MRI) in 61 elders selected for elevated vascular risk factors [41]. Autopsy studies of patients with AD suggest that mixed pathology (vascular disease, and alpha-synucleinopathies) is not unusual [42-44]. In a study of 116 elderly subjects followed longitudinally to autopsy, vascular lesions in brain of subjects with AD also contributed to loss of cortical gray matter [45]. Likely the inflammation of endothelial dysfunction that accompanies vascular disease adds an inflammatory burden to CNS-specific inflammatory lesions in AD patients.
Omega-3 (ω3) fatty acids participate in inflammatory balance at the vascular endothelium, as elsewhere in the brain. Docosahexaenoic acid (DHA) is critical for integrity of the cerebrovascular endothelium, a critical component of the BBB (Figure 1). Eicosapentaenoic acid (EPA) induces assembly of tight junctions in brain capillary endothelial cells [46]. Mouse brain micro vessel endothelial cells contain EPA and produce eicosanoids including PGI2 and PGE2, and this production is reduced on exposure to more EPA. EPA and DHA are transported across the human endothelial cell-brain barrier by diffusion in free and albumin-bound forms [47], and transported by the major facilitator super family domain (MSFD2a) when conjugated with lysophosphatidylcholine [48]. The decrease of free DHA that we see in CSF in early and late stages of AD (Table 1 & Figure 3) is consistent with a loss of this source of the neuroprotective resolvins. The protective anti-inflammatory effects of ω3 fatty acids (particularly phosphatidylcholine docosahexaenoic acid or DHA) are well known for cardiovascular health, and are likely equally important for brain health.
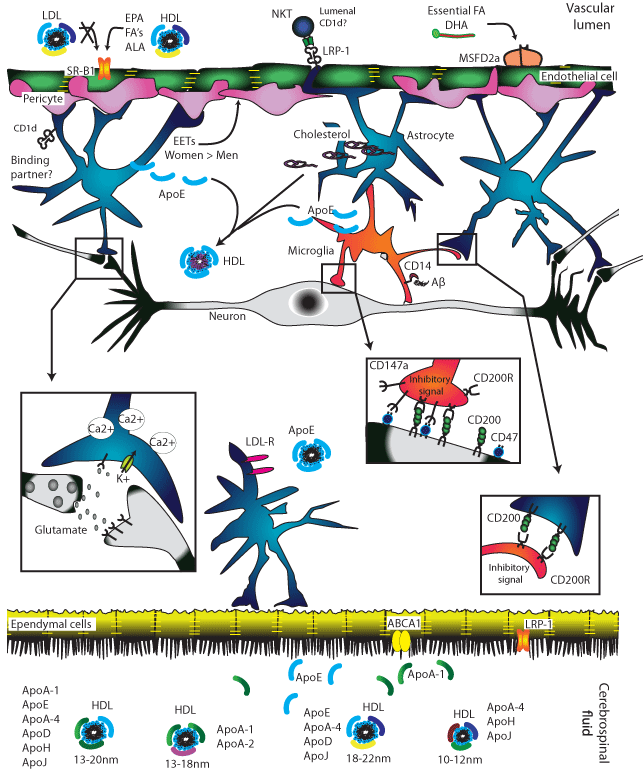
Figure 1: The interaction of several cell types is necessary to support neuronal function.
Starting top left clock wise. Brain micro vessels are lined with endothelial cells that form tight junctions (yellow bars) and are partially covered with pericytes
connected to astrocyte end-feet. These three cell types form the BBB [141]. Astrocytes express CD1d, a non-classical MHC molecule capable of presenting nonclassical
peptide and lipid antigen [142-143]. When the BBB is perturbed, astrocytes may nudge into the tight junctions and pericyte CD1d may even reach into the
vascular lumen, attracting cells from the peripheral blood (including NK and T cells). The binding partner for CD1d expressed in the brain parenchyma is unknown.
The BBB segregates cholesterol produced by astrocyte from cholesterol in the systemic circulation. Nearly all of the cholesterol in brain is synthesized in the
brain [115]. Blood transports lipids in the form of HDL, LDL, and VLDL particles but, of these, only small HDL particles cross the endothelium, probably using
the scavenger receptor class B type I (SR-BI) receptor [115]. Lipoproteins involved in lipid transport do not cross the normal BBB, and are mainly synthesized
in astrocytes and microglia [115]. The essential fatty acid DHA is exclusively transported as a lysophosphatidylcholine (LPC) in a sodium-dependent matter via
transcytosis with the major facilitator superfamily domain-containing protein 2a (MFSD2a), found only in brain microvessel endothelium [48].
Cholesterol is coated with astrocyte or microglia-derived ApoE to form HDL particles for intracellular trafficking [144]. Microglias (orange cells) is the local resident
macrophage population and as such, scavenge cellular debris and protein from the interstitiumincludingc tangles. Microglial phagocytosis of Aβ occurs via binding
of Aβ to microglial CD14 [145]. Microglia receives tonic inhibitory signals from both astrocytes and neurons via direct contact: CD147a and CD200R are expressed
by the microglia and neurons express the ligands for these receptors. Astrocytes only express the CD200R ligand [146-149].
CSF of healthy adults contains about 1 billion ‘nanoparticles’ per ml [12]. Abundant lipoprotein particles contain a variety of fatty acids (as indicated) but LDL is
generally not a component of these particles in brain, whereas HDL is [150].
Astrocytes (blue cells) scavenge neurotransmitters spilled over from neuronal synapses as part of the “tripartite synapse“to maintain healthy electrical signaling
[151]. Astrocytes are also involved in clearing plaque.
Analysis of the Framingham Heart Study revealed that higher blood plasma levels of DHA, the brain-enriched ω3 fatty acid, are associated with reduced risk of all-cause dementias [49]. Treatments aimed at boosting DHA to prevent or treat AD have had mixed results. For instance, DHA dietary supplements, shown to cross the blood-brain-barrier [50], reduced pro-inflammatory ω6 levels and had a positive effect on anti-inflammatory DHA levels [51]. However, a randomized trial of DHA supplementation did not have a clear beneficial clinical effect [52]. DHA which plays an important role in early brain development [53] and in reducing coronary heart disease [54] certainly deserves further study in the context of AD, despite initial ‘negative’ trials.
The abundant saturated fatty acids also influence the vascular endothelium. Cap rate has been shown to open the blood brain barrier [55] and, at 5-30 mM at the cellular level, opens the paracellular space in brain endothelial cells by acting on the tight junction claudin-5 and the actin skeleton [56]. Palmitate is toxic to porcine cerebromicrovascular endothelial cells [57]. Palmitoylation is a reversible post-translational modification that affects many brain capillary membrane proteins, such as the beta-adrenergic receptor and various G-proteins involved in signal transduction, vesicular transport, and cell differentiation [58]. Palmitoylated GLUT-1 is reportedly increased in hyperglycemic states [59]. Our CSF findings showing loss of protective ω3 and increase of harmful ω6 [7], combined with a known negative effect with this pattern of expression on endothelial function suggests that protective ω3 still deserves study as a therapy for delay or treatment of AD, and many such clinical studies are ongoing.
The loss of integrity of the BBB may allow chronic systemic inflammation to exacerbate neurodegeneration. In particular C-reactive protein (CRP) elevations in blood are correlated with cognitive decline [60]. Receptor for advanced glycation end-products (RAGE) elevation is also associated with cognitive impairment [61]. Together these observations suggest that the role of the peripheral immune system in AD may be underestimated. Nonetheless the peripheral immune system is represented by relatively small numbers of cells in the brain, so that the CSF lipids analyzed in our studies came from CNS cells, including all non-neuronal cells that constitute the innate immune system of the brain. The major populations of CNS immune cells are glia, but pericytes and mesenchymal cells are also part of the CNS immune response: Pericytes, one of the main cellular components of CNS BBB (along with astrocyte end-feet, and endothelial cells) certainly play a role in damping inflammation by limiting toxin entry into the brain via their mesangial function (Figures 1-3). Pericytes are a heterogeneous population of cells, and can acquire a microglial phenotype after stroke, and migrate away from their perivascular niche [62]. Pericytes can assume a macrophage (phagocytic) phenotype but whether pericytes operate as phagocytes in human AD is unknown. In bovine cell culture models, amyloid Aβ fragments found in AD brains induce phosphorylation of cytosolic phospholipase A2 (cPLA2) and activated cPLA2 generates pro-inflammatory arachadonic acid [63,64]. Aβ is toxic to pericytes, leading Zlokovic and colleagues to speculate that toxic loss of BBB pericytes from Aβ leads to a vicious cycle increasing Aβ deposition in brain and more pericyte loss [65].
Mesenchymal stem cells (MSC) or mesenchymal stromal cells are being tested in numerous clinical trials to reduce inflammatory processes in a variety of neurologic diseases including stroke, cerebral palsy, traumatic brain injury, inherited CNS diseases, multiple sclerosis, and Alzheimer’s disease. (See clinicaltrials.gov for more detail). In these clinical trials the sources of mesenchymal stem cells are bone marrow, adipose, or umbilical cord. In vitro, mesenchymal stem cells can alter the phenotype of activated microglia, inducing microglial expression of neuroprotective factors [66]. So though MSC are generally thought of as anti-inflammatory in other organ systems, in brain they may play a protective role by secretion of trophic factors that protect neural stem cells [67]. Little is known about the resident MSC population of the human brain because it is not yet possible to study resident MSC in intact brain, the only MSC isolated from human brain have been from tumors. The perivascular niche of human brain contains a mesenchymal stem cell population that expresses both MSC and pericyte markers, and in clonal assays, differentiates along both mesodermal and neurectodermal lineages [68].
Mesenchymal stem cells generally home to sites of inflammation but there is no published evidence that investigators have looked for MSC in AD lesions. From thousands of studies and from early clinical trials, MSC have profound immunomodulatory effects on T cells and peripheral macrophages, with potent (though probably transient) anti-inflammatory effects after transplantation [69]. The interaction of resident MSC in brain with the resident immune cells deserves further exploration in the context of AD. Inflammatory environments ‘activate’ MSC to interact with immune cells and turn down the proinflammatory traits of these cells, and toll-like receptors (TLRs) may be involved in this activation (though this is controversial and likely varies by MSC subtype [70].
Microglia and Astrocytes Constitute the Innate Immune System of the Brain
Microglia and astrocytes are both heterogeneous populations, analogous to heterogeneity of peripheral myeloid cell populations (Figures 1 & 2). Microglia constitutes about 10% of cells in the brain [71]. Microglia accumulates in areas of the brain undergoing neurodegeneration, and microglial activity causes release of proinflammatory molecules including reactive oxygen species (ROS) and nitric oxide. Depending on the initiating signal, activated microglia upregulate chemokine receptors, a large variety of cytokine receptors, complement receptors, lectin receptors and prostaglandin receptors as well as anti-inflammatory mediators such as IL-10 and PGE2 [72]. Microglia also serves as the macrophages of the brain, reflecting their role in development to prune neuronal populations [73]. Like peripheral macrophages, microglia express TLRs specialized for the sensing of danger signals, emphasizing their role in surveillance of the brain environment [74], (Figures 1 & 2).
Microglia in the healthy aged brain develops a “dystrophic” phenotype, characterized by an enlarged cell body, deramification or fragmentation of cellular extensions, and various cytoplasmic morphologic abnormalities [75]. Dystrophic microglia also express high levels of ferritin, an iron storage protein, making them potentially more susceptible to the oxidized microenvironment in which lipid peroxidation (prominent in our CSF results) is progressing [76]. Microglial mitochondria are considered highly vulnerable for age-accumulated DNA damage from ROS production [30,77]. Not surprisingly, the autophagic function of microglia is also impaired with aging [78]. Together the reduced functional capacities of microglia with aging may be important in setting up susceptibility to neurodegenerative changes of AD [73]. The activated phenotype of AD can be thought of as an extension of the dystrophic phenotype of aging. This activation is not all-or-none and manipulation of the phenotype of microglia into a less inflammatory state is a potential novel therapeutic approach to neurodegenerative diseases [66].
Amyloid plaque deposits in pathologic AD brains are often surrounded by microglia, and microglia are able to clear plaques early in the course of AD, leading to suggestions that increasing numbers of microglia could also be therapeutic in AD. Microglial CD14 (a coreceptor with TLR4 for lipopolysaccharide) interacts with fibrillary Aβ, promoting its phagocytosis [79], but phagocytosis of microglia is impaired in AD (Figure 1 and 2), [80]. Murine studies demonstrate that microglia change their normal secretory profile and release proinflammatory cytokines in response to Aβ deposition in later stages of AD [81,82]. Once large deposits of plaque are present in the brain, microglia may simply become overwhelmed, exacerbated by their senescence (reduced function) phenotype.
Lipids in Neurodegenerating Sites Can Act as Chemo attractants for Microglia
Recently, a direct microglia-lipid interaction was unearthed that is clearly critical to normal function of microglia in response to toxic Aβ. The microglial surface receptor TREM2 (for triggering receptor expressed on myeloid cells-2) directly senses lipids associated with Aβ in areas of neurodegeneration (Figure 2), leading to a model in which lipids (not just Aβ) exposed by degenerating neurons and astrocytes may be the activating cues for microglia [83]. Phosphatidylcholine (PC) and sphingomyelin (SM) are particularly potent inducers of TREM2 activity in experimental models but membrane phospholipids such as phosphatidylserine can also induce TREM receptor activation [83]. These studies point to a powerful feed-forward lipid loop for microglia in the context of AD: Lipids attract dysfunctional (aged) microglia in to a toxic neurodegenerating site, where other oxidized lipids contribute further to microglia demise. TREM2 heterozygosity is associated with a significant risk of AD [84]. Loss of function studies of TREM2 in mouse models of AD lead to contradictory conclusions about the role of TREM2 in in AD, but clearly point to the importance of studying TREM2 action in microglia as a potential approach to AD therapy [85].
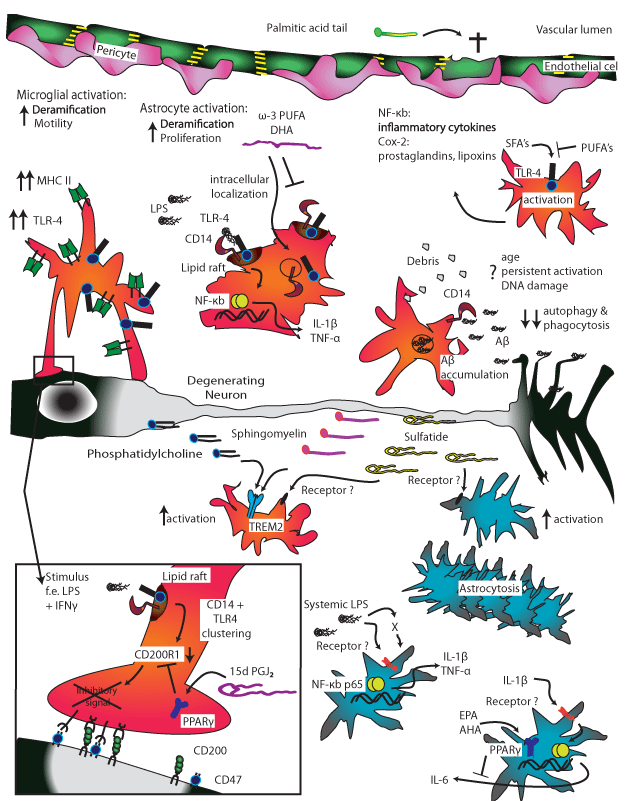
Figure 2: The supportive network of brain innate immune cells reacts to diverse inflammatory stimuli.
Starting top left clockwise. Microglia is the resident macrophage population of the brain [152-153]. Microglia phagocytose and remove extracellular debris, misfolded
proteins and apoptotic cells that are toxic to neurons [153]. With age, microglial phagocytic capacity decreases, whether this is due to chronic activation or DNA
damage is unclear. Aβ tangles can accumulate in microglia that seemingly lacks the ability to digest these complex proteins [154]. Reduced ability to clear plaque
debris likely contributes to neurotoxicity even early in AD [155].
Microglial activation and changes with age include deramification (loss of cellular processes), increased motility and increased expression of MHC II and TLR-4
[75,156,157]. TLR-4 and CD14, known to bind LPS, are also involved in uptake of Aβ. Ligand binding at these sites induces activation in the lipid bilayer [158]. This
activation induces the relocation of NF-kB to the nucleus where expression of proinflammatory genes such as IL-1β and TNFα are upregulated. Eicosapentaenoic
acid blocks LPS-mediated activation of microglia via an unknown mechanism [159]. Omega-3 polyunsaturated fatty acids (PUFA) such as DHA and EPA prevent
surface location of the receptors and thus inhibit TLR-4-mediated activation [160].
A typical morphological feature of AD is astrocytosis, or astrocyte proliferation [161].
Stressed neurons release sphingomyelin and phosphatidylcholine which binds TREM2 receptors on microglia inducing activation [83]. Sulfatide, a major component
of myelin sheaths, similarly induces activation and release of NO, TNFα, IL-6 and IL-12 in microglia and astrocytes [162]. The proinflammatory cytokine IL-1β
induces IL-6 expression in astrocytes, an effect that can be inhibited by EPA, DHA, or ALA via the transcription factor PPAR-γ [163]. Saturated fatty acids bind
TLR-4 and result in NF-κB-mediated gene expression [164]. PUFAs inhibit this TLR-4 mediated activation in a dominant fashion [165]. TLR-4 sequence variants
are associated with risk of late onset Alzheimer’s disease (LOAD), especially in ApoE e4 non-carriers [166].
The brain innate immune system is quiescent in healthy brains. Stimulus with inflammatory agents such as IFNγ and LPS that bind to CD14+TLR-4 induce
clustering of these receptors within the lipid rafts. The proximity enables signal transduction and results in the downregulation of CD200R1 [167]. In absence of
surface CD200R1, the inhibitory signal cannot be delivered and default microglial activation takes place. Presence of the resolving 15dPGJ2, which binds to the
transcription factor PPARγ dominantly, inhibits the downregulation of CD200R1 in the presence of inflammatory stimuli, thus preventing microglial activation [167].
Microglia process Aβ by engulfing cells and by taking up exosomes that contain Aβ. Exosomes are small membrane vesicles released via exocytose is by many cell types. Exosomes are now recognized as important pathways of cell-cell communication [86] in which exosomalnucleic acids, proteins and lipids function as signaling molecules [87]. In CSF of our AD subjects, Aβ levels are reduced. This finding is consistent with other reports [88] and the CSF Aβ levels correlate inversely with amyloid plaque load in the AD brain [89]. Reiterating this important point: As disease progresses Aβ levels in CSF fall. These findings may suggest that the normal clearance of Aβ through CSF is damaged or over whelmed as AD progresses, but the mechanisms responsible for processing amyloid precursor protein and trafficking of the downstream Aβ products are not fully elucidated [90,91].
Secretion of exosomes containing Aβ is under control of sphingolipid-metabolizing enzymes (sphingomyelinases) [92] and exosomes accumulate in the plaque of AD brains [93]. In the CSF of AD patients we found significantly reduced acid sphingomyelinase [8] , which may potentially contribute to the inability of microglia to clear Aβ in the exosome compartment of the brain. Others have reported increased acid sphingomyelinase activity in brains of postmortem AD subjects [94], a difference which may be due to the brain compartments studied or stage of disease. Given that Aβ levels decrease in CSF of AD subjects, the reduced acid sphingomyelinase activity that we saw may contribute to reduced trafficking or clearance of Aβ into CSF. More generally our results suggest that the normal machinery used by Golgi and lysosomes to generate endosomes and phagosomes, as well as cellular exosome formation, content, and exocytosis are all disturbed by loss of acid sphingomyelinase activity in AD brains [8].
Sphingolipids are central regulators of cell growth, differentiation and death, and so changes in sphingolipid content in CSF of AD subjects represent important findings [8]. Previous work has emphasized increases in the sphingolipid metabolites, ceramides, in AD brains [95] as potentially toxic and promoting neuronal death and inhibiting autophagy [96]. These studies emphasize the importance of quantifying all members of these complex lipids in the context of AD, in order to more fully evaluate their role in pathogenesis, as sphingolipids balance pro- and anti-inflammatory functions [97,98].
The discrepancy between loss of acid sphingomyelinase activity in CSF and increased ceramide accumulation in brains of AD subjects also deserves comment. Like the inverse correlation between low Aβ in CSF and high brain levels with progression of AD, the low levels of acid sphingomyelinase in CSF do not necessarily reflect the levels in brain. The normal partitioning and trafficking of the enzyme may be disturbed with disease progression, but more intensive study of CSF over time [89] will be required to document the flux of acid sphingomyelinase in CSF. The abundance of ceramide in the brain depends on the balance between its synthesis, degradation/metabolism and compartmentalization, the subject of a recent computational model [99]. In addition to various sphingomyelinases in different anatomic and cellular compartments, ceramideabundance is regulated by a de novo synthesis pathway and by hydrolysis of ceramidases. In our published work [8] we showed that ceramide decreased in CSF nanoparticles but was increased in the supernatant compartment of CSF. Acid sphingomyelinase is essential for maintaining proper cellular concentrations of sphingolipids [100], and so the reduced ceramide we find in the nanoparticles (which likely include lysosomes) is consistent with the loss of acid sphingomyelinase in the nanoparticle fraction. The increased ceramide in the CSF soluble fraction is probably from de novo synthesis, a source of ceramide under control of the orosomucoid-like gene family [101].
Given the centrality of phagosomes in antigen presentation in the periphery [102], and the immune function of exosomes in many organ systems, the disruption of acid sphingomyelinase signaling in AD likely effects multiple levels of immune function in AD. Furthermore exosomes can cross the BBB and influence any peripheral organ including the immune system, and are currently being studied as carriers of therapeutic cargo [103]. The pathologic implications for immune function in response to brain exosomes carrying abnormal cargo are completely unexplored.
It is important to note that our CSF lipid studies were performed separately on the (i) soluble fraction of CSF which reflects brain interstitium and on the abundant (ii) CSF particles which we call ‘nanoparticles’ [8] (Figure 3). Our characterization of the nanoparticles is far from complete but they certainly containexosomes, microsomes, as well as shed cellular debris [12]. In our results lipid changes in one of these CSF compartments are often different from changes in the other, highlighting the importance of studying both. We do not know the cellular source of these particles (most groups ignore them when analyzing CSF), or how much of this particle fraction is exosomes. Some of the particles are morphologically consistent with synaptic vesicles by immuno-electron microscopy [12]. In normal CSF we find ~1 billion particles/mL and the number of particles increases in AD patients (unpublished). Given that normal exosome formation is likely disturbed in AD, the increased numbers of nanoparticles as disease progresses likely reflects cellular debris accompanying neurodegeneration.
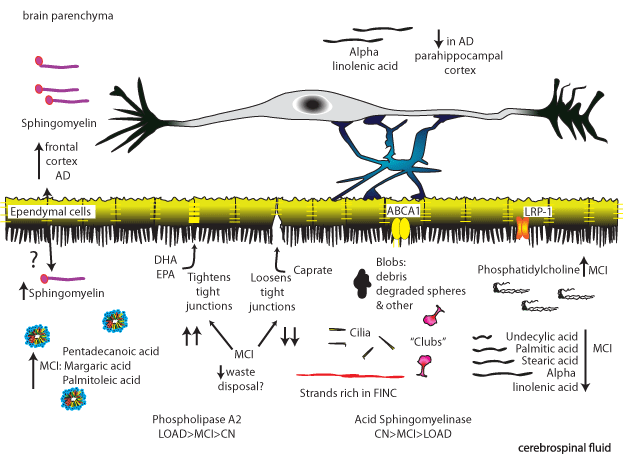
Figure 3: The interplay of blood, brain parenchyma and CSF.
Starting on the top right moving clockwise (in the CSF). DHA and EPA induce loosening of tight junctions while caprate induces tightening [46,55]. Cap rate is
reduced in MCI while DHA and EPA are increased, possibly resulting in overall tighter tight junctions and an inability to dispose of waste [7].
In CSF from MCI subjects, the content of pentadecanoic acid, margaric acid, and palmitoleic acid in CSF particles increases [12].
CNS levels of ceramide are increased in AD and the lipid can also be found in increased amounts in frontal cortex astrocytes of AD patients [168].
Different types of debris can be detected in CSF, even in healthy adults. Gross morphology includes blobs with non-specific debris, and degraded spheres.“Clubs”
are likely dislodged structures from the ventricle wall and broken off cilia as well as strands that form a CSF matrix rich in fibronectin [12].
See text for changes in CSF content of fatty acids, phospholipase A2 and acid sphingomyelinase MCI and LOAD [6,8,12]. Anti-inflammatory alpha-linoleic acid
content decreases in the parahippocampal cortex of AD patients [169].
Microglia not only process exosomes, they release their own ‘microvesicles’ that contain inflammatory mediators such as TNF-α and IL-1β [104,105]. The interaction between microglia and astrocytes controls microglial shedding of microsomes [106].
Candidate microglial mediators that promote inflammation in AD were recently identified from a whole-genome sequencing study of subjects with presenilin-1 mutations (a familial early onset form of AD). In the examined cohort, age of clinical disease onset is quite predictable. Subjects with delayed age of onset were found to have mutations in a gene cluster coding for several cytokines [107]. In particular a single nucleotide polymorphism in CCL11, encoding eotaxin-1, was associated with delayed age of onset. Eotaxin-1 increases with age and high levels are correlated with impaired neurogenesis. Eotaxin-1 binds the CCR3 receptor on microglia. This work also implicated IL-4 and CCL2 (a pro-inflammatory cytokine) as potential modulators of age of disease onset, and so potential targets for AD therapies. IL4 receptor is expressed on microglia, and IL4 (with IL13) signaling is involved in clearance of Aβ [107].
Astrocytes are by far and away the most abundant cells in brain, playing critical roles in CNS ion balance, synaptic remodeling, and regulation of energy balance [108]. Astrocytes can uptake glucose from the circulation, store glycogen or generate lactate to meet the neuronal energy demand [109]. Astrocytes are involved in homeostasis of extracellular space; they can express potassium channels and contribute to clearing extracellular potassium or glutamate around synapses [110]. In addition to supplying metabolic substrates for neurons, astrocyte endfeet ensheath blood vessels, contributing to BBB structure and control of cerebral blood flow [28,108]. The ratio of astrocytes to neurons is much higher in primates than rodents, suggesting that astrocytes may be more important in maintaining brain homeostasis in humans than in lower mammals, and even in higher order functioning (cognition) of primates vs. rodents [111].
In human Alzheimer’s disease, activated astrocytes express myeloperoxidase, and in experimental AD models myeloperoxidase preferentially (per) oxidizes lipids. Myeloperoxidase, normally expressed in neutrophils, is not expressed in healthy brains [112]. Reports of phosphatidylserine loss in erythrocytes in AD subjects generated some enthusiasm for replacement therapy as a potential way to support the brain of these patients [113,114]. We also observed a loss of phosphatidylserine in CSF in early AD subjects (before memory impairment) vs. in CSF of healthy age-matched controls (See Table 1, Figure 3). Although replacement of phosphatidylserine represents a logical therapeutic approach to AD, clinical studies to date have not shown a dramatic improvement in cognition with its supplementation [113]. The lack of strong therapeutic signal in these studies may be the result of starting supplementation too late in the disease process, or use of only a single supplement, or poor bioavailability of current formulations.
Only some small high density lipoproteins (HDL) made in the liver cross the BBB [115]. Nonetheless a pattern of higher LDL cholesterol and lower HDL cholesterol (the pattern considered high risk for cardiovascular disease) is associated with higher levels of cerebral amyloidosis by Compound B positron emission tomography [116]. In the brain astrocytes synthesize and provide the majority of the apolipoproteins (including ApoE) and cholesterol needed by neurons [117]. Astrocytes use ApoE-containing lipoproteins to control cholesterol distribution (Figure 1). Exposure of primary rat astrocytes to CSF from AD (or other dementia) subjects resulted in reduced cholesterol efflux from the astrocytes [118], and suggests that products secreted in the AD diseased environment damage astrocyte functions important to brain health.
Though not of hematopoietic origin, astrocytes are key immune modulators. They function as antigen presenting cells and as a barrier to influx of peripheral immune cells into the brain. Connexin-43 on astrocytes at the BBB is critical to BBB function [119]. Astrocytes are also the source of neurotrophins, so that disrupted function of astrocytes by inflammation leads to loss of factors that support neuron survival. Like microglia, stimulated astrocytes become activated and change their morphology as well as expression profiles, and in AD, migrate to sites of Aβ deposition. In in vitro studies conditioned medium from activated (IFN-γ stimulated) human astrocytes is neurotoxic rather than neurotrophic [120]. Like microglia, the initially protective activated astrocytes transition to a neuronaldamaging phenotype through both secretion of harmful molecules, and loss of neuroprotective functions [121] with disease progression. Surprisingly little work utilizes primary human astrocytes to study AD, a lost opportunity for understanding the interplay of astrocytes and immune function in the context of this disease. Magnetic resonance spectroscopy suggests glial activation or inflammation very early in human AD, before the onset of cognitive impairment [122]. This early glial activation in the course of AD is supported by recent PET studies in mice, underlining the need to understand the contribution of non-neuronal brain cells in the earliest stages of disease [123].
Initiation of immune responses is a well-controlled process. The inflammasome is a multiprotein complex that initiates a powerful inflammatory cascade. The inflammasome is comprised of an assembly of ‘sensory’ proteins, adapter proteins, and caspases that cleave inflammatory proteins (such as IL-1β) from their pro-forms into active forms, triggering the inflammatory response. In the CNS microglia, neurons, and astrocytes express different components of the inflammasome complex. Human astrocytes express NLR protein 2 which is activated by danger-associated molecular pattern (DAMP) ATP, and ultimately leads to processing of caspase-1 and interleukin- 1β [124]. Importantly, polymorphisms of the major inflammatory protein generated by inflammasome activation, IL-1β, is associated with age at AD onset [125]. Among the many signals that can incite or quell inflammasome activity, fatty acids [126,127] are of special interest in the context of our data. Most work on inflammasomes has been done in peripheral organ systems, pointing to another important opportunity for use of primary human astrocytes (or iPSderived astrocytes) to understand the initiation and perpetuation of inflammation in the setting of AD.
Like microglia, astrocyte immune function in the brain involves phagocytosis (and therefore clearing) of Aβ [128,129]. In vitro studies suggest that Aβ exposure induces increased expression of CD36 and CD47 (which may recognize Aβ) on astrocytes, but receptor for advanced glycationend-products (RAGE) expression is reduced [130]. As is the case for vascular disease, advanced glycation end- (AGE) products accumulate with amyloid in AD, and contribute to increased oxidant stress. AGE may directly induce lipid peroxidation [131], and such (per) oxidized lipids are prominent in CSF of our AD subjects.
Phospholipids are the major source of several lipid mediators of inflammation. Polyunsaturated fatty acids (PUFAs) such as arachidonic acid (20: 4, n-6) or DHA are released from membranebound phospholipids by phospholipases and metabolized in multistep enzymatic pathways to generate signaling molecules, collectively known as eicosanoids. A striking finding in our CSF studies is the high levels of PLA2 in AD subjects [6]. An increase in PLA2 will not only initiate the formation of arachidonic acid-derived lipid mediators of inflammation [10,132], but may disrupt membrane structure sufficient to result in abnormal processing of amyloid precursor proteins. See Figure 4.
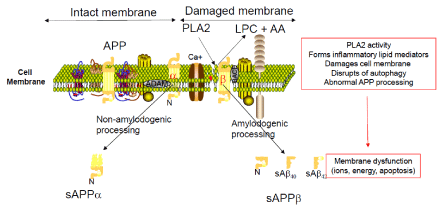
Figure 4: PLA2 activity damages membranes.
Amyloid precursor proteins (APP) processed through the non-amyloidogenic pathway by alpha secretases generate nontoxic soluble peptide fragments Macias
et al, 2014; Zhao et al, 2014 [170]. Increased PLA2 activity in AD Emmerling et al, 1993 [6] may initiate the formation of lipid mediators of inflammation in addition
to damaging the cell membranes structurally. Damaged cellular membranes may expose APP to abnormal processing by beta secretases Grimm et al, 2012;
Bhattacharyya et al, 2013;Lemmin et al, 2014, [171] resulting in formation of neurotoxic peptide fragments that aggregate to form plaques. Damaged membranes
may also disrupt ion channels and ion gradients, resulting in dysfunctional neuronal repair. These processes contribute to neurodegeneration in AD.
Polyunsaturated fatty acids are converted to potent inflammatory mediators by prostaglandin synthases, lipoxygenases, or CP450 enzymes in conjunction with PLA2 isoforms; some of these enzymes are altered in AD [133-135].
Prostaglandins and leukotrienes are the major eicosanoids synthesized by the cyclooxygenase and lipoxygenase pathways, respectively. Several studies have documented changes in the eicosanoid pathway in AD [136-139]. Given that eicosanoids have pleiotropic bioactive properties– in inflammation, the immune response, platelet aggregation, chemotaxis, vasodilation, and ion channel function, and cell growth and differentiation patterns, modulation of eicosanoid synthesis may have therapeutic potential in AD [138,140].
Conclusions
We used our database of CSF lipids from an ongoing AD clinical study as a platform to highlight the importance of interactions between lipid metabolism and inflammation and the immune system in AD. Like other groups, we show that anti-inflammatory lipids (DHA) known to play a role in vascular and brain health are significantly reduced in CSF of subjects with early AD. Abnormal, high levels of many inflammatory lipids in CSF of these subjects are prominent in our data. Our results suggest that the changes in lipids we see can negatively impact immune functions of microglia and astrocytes, as well as brain vascular health, beyond the well-known toxic effects of amyloid plaques. The extensive changes of CSF lipids we reported recently, predominantly in brain-derived nanoparticles, indicate that compromised brain cell membrane lipid composition is a major part of early AD pathology. We hope this discussion of specific pathophysiologic effects of lipids will lead to a better understanding of AD.
References
- Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, et al. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006; 12: 138-143.
- Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. 2015; 16: 229-236.
- Sultana R, Perluigi M, Allan Butterfield D. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med. 2013; 62: 157-169.
- Leonarduzzi G, Gamba P, Gargiulo S, Biasi F, Poli G. Inflammation-related gene expression by lipid oxidation-derived products in the progression of atherosclerosis. Free Radic Biol Med. 2012; 52: 19-34.
- Yang X, Askarova S, Lee JC. Membrane biophysics and mechanics in Alzheimer's disease. Mol Neurobiol. 2010; 41: 138-148.
- Fonteh AN, Chiang J, Cipolla M, Hale J, Diallo F, Chirino A, et al. Alterations in cerebrospinal fluid glycerophospholipids and phospholipase A2 activity in Alzheimer's disease. J Lipid Res. 2013; 54: 2884-2897.
- Fonteh AN, Cipolla M, Chiang J, Arakaki X, Harrington MG. Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimer's disease. PLoS One. 2014; 9: e100519.
- Fonteh AN, Ormseth C, Chiang J, Cipolla M, Arakaki X, Harrington MG. Sphingolipid metabolism correlates with cerebrospinal fluid Beta amyloid levels in Alzheimer's disease. PLoS One. 2015; 10: e0125597.
- Harrington MG, Chiang J, Pogoda JM, Gomez M, Thomas K, Marion SD, et al. Executive function changes before memory in preclinical Alzheimer's pathology: a prospective, cross-sectional, case control study. PLoS One. 2013; 8: e79378.
- Fonteh AN, Harrington RJ, Huhmer AF, Biringer RG, Riggins JN, Harrington MG. Identification of disease markers in human cerebrospinal fluid using lipidomic and proteomic methods. Dis Markers. 2006; 22: 39-64.
- Fonteh AN, Fisher RD. Combining lipidomics and proteomics of human cerebrospinal fluids. Methods Mol Biol. 2009; 579: 71-86.
- Harrington MG, Fonteh AN, Oborina E, Liao P, Cowan RP, McComb G, et al. The morphology and biochemistry of nanostructures provide evidence for synthesis and signaling functions in human cerebrospinal fluid. Cerebrospinal Fluid Res. 2009; 6: 10.
- Greenwood J, Etienne-Manneville S, Adamson P, Couraud PO. Lymphocyte migration into the central nervous system: implication of ICAM-1 signalling at the blood-brain barrier. Vascul Pharmacol. 2002; 38: 315-322.
- Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH . An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. 2007; 204: 2023-2030.
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007a; 28: 12-18.
- Ransohoff RM, Kivisäkk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003; 3: 569-581.
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015; 523: 337-341.
- Xie L, Choudhury GR, Winters A, Yang SH, Jin K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur J Immunol. 2015; 45: 180-191.
- Sankowski R, Mader S, Valdés-Ferrer SI. Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosci. 2015; 9: 28.
- Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006; 213: 48-65.
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015; 85: 296-302.
- Bargerstock E, Puvenna V, Iffland P, Falcone T, Hossain M, Vetter S, et al. Is peripheral immunity regulated by blood-brain barrier permeability changes? PLoS One. 2014; 9: e101477.
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009; 118: 103-113.
- Mabuchi T, Lucero J, Feng A, Koziol JA, del Zoppo GJ. Focal cerebral ischemia preferentially affects neurons distant from their neighboring microvessels. J Cereb Blood Flow Metab. 2005; 25: 257-266.
- Tsai PS, Kaufhold JP, Blinder P, Friedman B, Drew PJ, Karten HJ, et al. Correlations of neuronal and microvascular densities in murine cortex revealed by direct counting and colocalization of nuclei and vessels. J Neurosci. 2009; 29: 14553-14570.
- Jaspersen SL, Ghosh S. Nuclear envelope insertion of spindle pole bodies and nuclear pore complexes. Nucleus. 2012; 3: 226-236.
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006; 7: 41-53.
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010; 468: 232-243.
- Kroetz DL, Xu F. Regulation and inhibition of arachidonic acid omega-hydroxylases and 20-HETE formation. Annu Rev Pharmacol Toxicol. 2005; 45: 413-438.
- Shi Y, Liu X, Gebremedhin D, Falck JR, Harder DR, Koehler RC. Interaction of mechanisms involving epoxyeicosatrienoic acids, adenosine receptors, and metabotropic glutamate receptors in neurovascular coupling in rat whisker barrel cortex. J Cereb Blood Flow Metab. 2008; 28: 111-125.
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008; 456: 745-749.
- Lecrux C, Toussay X, Kocharyan A, Fernandes P, Neupane S, Lévesque M, et al. Pyramidal neurons are "neurogenic hubs" in the neurovascular coupling response to whisker stimulation. J Neurosci. 2011; 31: 9836-9847.
- Canobbio I, Abubaker AA, Visconte C, Torti M, Pula G. Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer's disease. Front Cell Neurosci. 2015; 9: 65.
- Takeda S, Sato N, Morishita R. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: relevance to pathogenesis and therapy. Front Aging Neurosci. 2014; 6: 171.
- Stranahan AM, Martin B, Maudsley S. Anti-inflammatory effects of physical activity in relationship to improved cognitive status in humans and mouse models of Alzheimer's disease. Curr Alzheimer Res. 2012; 9: 86-92.
- Medhora M, Narayanan J, Harder D. Dual regulation of the cerebral microvasculature by epoxyeicosatrienoic acids. Trends Cardiovasc Med. 2001; 11: 38-42.
- Medhora M, Narayanan J, Harder D, Maier KG. Identifying endothelium-derived hyperpolarizing factor: recent approaches to assay the role of epoxyeicosatrienoic acids. Jpn J Pharmaco. 2001a; l86: 369-375.
- Zhang C, Harder DR. Cerebral capillary endothelial cell mitogenesis and morphogenesis induced by astrocytic epoxyeicosatrienoic Acid. Stroke. 2002; 33: 2957-2964.
- Carver KA, Lourim D, Tryba AK, Harder DR. Rhythmic expression of cytochrome P450 epoxygenases CYP4x1 and CYP2c11 in the rat brain and vasculature. Am J Physiol Cell Physiol. 2014; 307: C989-998.
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007; 69: 2197-2204.
- Marchant NL, Reed BR, Sanossian N, Madison CM, Kriger S, Dhada R, et al. The aging brain and cognition: contribution of vascular injury and aβ to mild cognitive dysfunction. JAMA Neurol. 2013; 70: 488-495.
- Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain. 2013; 136: 2697-2706.
- Toledo JB, Cairns NJ, Da X, Chen K, Carter D, Fleisher A, et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun. 2013; 1: 65.
- Arnold SE, Toledo JB, Appleby DH, Xie SX, Wang LS, Baek Y, et al. Comparative survey of the topographical distribution of signature molecular lesions in major neurodegenerative diseases. J Comp Neurol. 2013; 521: 4339-4355.
- Zheng L, Vinters HV, Mack WJ, Weiner MW, Chui HC. Differential effects of ischemic vascular disease and Alzheimer disease on brain atrophy and cognition. J Cereb Blood Flow Metab. 2015; .
- Yamagata K, Tagami M, Takenaga F, Yamori Y, Nara Y, Itoh S. Polyunsaturated fatty acids induce tight junctions to form in brain capillary endothelial cells. Neuroscience. 2003; 116: 649-656.
- Ouellet M, Emond V, Chen CT, Julien C, Bourasset F, Oddo S, et al. Diffusion of docosahexaenoic and eicosapentaenoic acids through the blood-brain barrier: An in situ cerebral perfusion study. Neurochem Int. 2009; 55: 476-482.
- Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X, et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature. 2014; 509: 503-506.
- Schaefer EJ, Bongard V, Beiser AS, Lamon-Fava S, Robins SJ, Au R, et al. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: the Framingham Heart Study. Arch Neurol. 2006; 63: 1545-1550.
- Freund Levi Y, Vedin I, Cederholm T, Basun H, Faxen Irving G, Eriksdotter M, et al. Transfer of omega-3 fatty acids across the blood-brain barrier after dietary supplementation with a docosahexaenoic acid-rich omega-3 fatty acid preparation in patients with Alzheimer's disease: the OmegAD study. J Intern Med. 2014; 275: 428-436.
- Wang X, Hjorth E, Vedin I, Eriksdotter M, Freund-Levi Y, Wahlund LO, et al. Effects of n-3 FA supplementation on the release of proresolving lipid mediators by blood mononuclear cells: the OmegAD study. J Lipid Res. 2015; 56: 674-681.
- Quinn JF, Raman R, Thomas RG, Yurko-Mauro K, Nelson EB, Van Dyck C, et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA. 2010; 304: 1903-1911.
- Auestad N, Scott DT, Janowsky JS, Jacobsen C, Carroll RE, Montalto MB, et al. Visual, cognitive, and language assessments at 39 months: a follow-up study of children fed formulas containing long-chain polyunsaturated fatty acids to 1 year of age. Pediatrics. 2003; 112: e177-183.
- Harris WS, Pottala JV, Lacey SM, Vasan RS, Larson MG, Robins SJ. Clinical correlates and heritability of erythrocyte eicosapentaenoic and docosahexaenoic acid content in the Framingham Heart Study. Atherosclerosis. 2012; 225: 425-431.
- Ohnishi T, Aida K, Awazu S. Enhancement of blood-brain barrier permeability by sodium caprate. J Pharm Pharmacol. 1999; 51: 1015-1018.
- Del Vecchio G, Tscheik C, Tenz K, Helms HC, Winkler L, Blasig R, et al. Sodium caprate transiently opens claudin-5-containing barriers at tight junctions of epithelial and endothelial cells. Mol Pharm. 2012; 9: 2523-2533.
- Ullen A, Fauler G, Bernhart E, Nusshold C, Reicher H, Leis HJ, et al. Phloretin ameliorates 2-chlorohexadecanal-mediated brain microvascular endothelial cell dysfunction in vitro. Free Radic Biol Med. 2012; 53: 1770-1781.
- Poulio JF, Béliveau R. Palmitoylation of brain capillary proteins. Int J Biochem Cell Biol. 1995; 27: 1133-1144.
- Pouliot JF, Béliveau R. Palmitoylation of the glucose transporter in blood-brain barrier capillaries. Biochim Biophys Acta. 1995; 1234: 191-196.
- Ravaglia G, Forti P, Maioli F, Montesi F, Rietti E, Pisacane N, et al. Risk factors for dementia: data from the Conselice study of brain aging. Arch Gerontol Geriatr. 2007; 44 Suppl 1: 311-320.
- Ge X, Xu XY, Feng CH, Wang Y, Li YL, Feng B. Relationships among serum C-reactive protein, receptor for advanced glycation products, metabolic dysfunction, and cognitive impairments. BMC Neurol. 2013; 13: 110.
- Özen I, Deierborg T, Miharada K, Padel T, Englund E, Genové G, et al. Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol. 2014; 128: 381-396.
- Anfuso CD, Assero G, Lupo G, Nicotra A, Cannavò G, Strosznajder RP, Rapisarda P. Amyloid beta(1-42) and its beta(25-35) fragment induce activation and membrane translocation of cytosolic phospholipase A2 in bovine retina capillary pericytes. Biochim Biophys Acta. 2004; 1686: 125-138.
- Nicotra A, Lupo G, Giurdanella G, Anfuso CD, Ragusa N, Tirolo C, et al. MAPKs mediate the activation of cytosolic phospholipase A2 by amyloid beta(25-35) peptide in bovine retina pericytes. Biochim Biophys Acta. 2005; 1733: 172-186.
- Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer's disease? Brain Pathol. 2014; 24: 371-386.
- Giunti D, Parodi B, Cordano C, Uccelli A, Kerlero de Rosbo N. Can we switch microglia's phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology. 2014; 141: 328-339.
- Salgado AJ, Sousa JC, Costa BM, Pires AO, Mateus-Pinheiro A, Teixeira FG, et al. Mesenchymal stem cells secretome as a modulator of the neurogenic niche: basic insights and therapeutic opportunities. Front Cell Neurosci. 2015; 9: 249.
- Paul G, Özen I, Christophersen NS, Reinbothe T, Bengzon J, Visse E, et al. The adult human brain harbors multipotent perivascular mesenchymal stem cells. PLoS One. 2012; 7: e35577.
- Eggenhofer E, Luk F, Dahlke MH, Hoogduijn MJ. The life and fate of mesenchymal stem cells. Front Immunol. 2014; 5: 148.
- Castro-Manrreza ME, Montesinos JJ. Immunoregulation by mesenchymal stem cells: biological aspects and clinical applications. J Immunol Res. 2015; 2015: 394917.
- Turola E, Furlan R, Bianco F, Matteoli M, Verderio C. Microglial microvesicle secretion and intercellular signaling. Front Physiol. 2012; 3: 149.
- Aloisi F. Immune function of microglia. Glia. 2001; 36: 165-179.
- Harry GJ. Microglia during development and aging. Pharmacol Ther. 2013; 139: 313-326.
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005; 308: 1314-1318.
- Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004; 45: 208-212.
- Brown DR. Role of microglia in age-related changes to the nervous system. ScientificWorldJournal. 2009; 9: 1061-1071.
- Hayashi Y, Yoshida M, Yamato M, Ide T, Wu Z, Ochi-Shindou M, et al. Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci. 2008; 28: 8624-8634.
- Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992; 48: 405-415.
- Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain. 2005; 128: 1778-1789.
- Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2010; 9: 156-167.
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007; 13: 432-438.
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008; 28: 8354-8360.
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015; 160: 1061-1071.
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013; 368: 117-127.
- Tanzi RE. TREM2 and Risk of Alzheimer's Disease--Friend or Foe? N Engl J Med. 2015; 372: 2564-2565.
- Frühbeis C, Fröhlich D, Kuo WP, Krämer-Albers EM. Extracellular vesicles as mediators of neuron-glia communication. Front Cell Neurosci. 2013; 7: 182.
- Mathivanan S, Fahner CJ, Reid GE, Simpson RJ. ExoCarta 2012: database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2012; 40: D1241-1244.
- Toledo JB, Zetterberg H, van Harten AC, Glodzik L, Martinez-Lage P, Bocchio-Chiavetto L, et al. Alzheimer's disease cerebrospinal fluid biomarker in cognitively normal subjects. Brain. 2015; 138: 2701-2715.
- Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG, et al. Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol. 2015; .
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011; 3: 89ra57.
- Ramanathan A, Nelson AR, Sagare AP, Zlokovic BV. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer's disease: the role, regulation and restoration of LRP1. Front Aging Neurosci. 2015; 7: 136.
- Yuyama K, Sun H, Mitsutake S, Igarashi Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J Biol Chem. 2012; 287: 10977-10989.
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, et al. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006; 103: 11172-11177.
- He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol Aging. 2010; 31: 398-408.
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A. 2004; 101: 2070-2075.
- Tamboli IY, Hampel H, Tien NT, Tolksdorf K, Breiden B, Mathews PM, et al. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Abeta generation. J Neurosci. 2011; 31: 1837-1849.
- Pettus BJ, Chalfant CE, Hannun YA. Sphingolipids in inflammation: roles and implications. Curr Mol Med. 2004; 4: 405-418.
- Nixon GF. Sphingolipids in inflammation: pathological implications and potential therapeutic targets. Br J Pharmacol. 2009; 158: 982-993.
- Wronowska W, Charzyńska A, Nienałtowski K, Gambin A. Computational modeling of sphingolipid metabolism. BMC Syst Biol. 2015; 9: 47.
- Jenkins RW1, Canals D, Hannun YA . Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. 2009; 21: 836-846.
- Kiefer K, Carreras-Sureda A, Garcia-Lopez R, Rubio-Moscardo F, Casas J, Fabrias G, et al. Coordinated regulation of the orosomucoid-like gene family expression controls de novo ceramide synthesis in mammalian cells. J Biol Chem. 2015; 290: 2822-2830
- Nair-Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, Huang Y, et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell. 2014; 158: 506-521.
- Tsilioni I, Panagiotidou S, Theoharides TC. Exosomes in neurologic and psychiatric disorders. Clin Ther. 2014; 36: 882-888.
- Antonucci F, Turola E, Riganti L, Caleo M, Gabrielli M, Perrotta C, et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 2012; 31: 1231-1240.
- Joshi P, Turola E, Ruiz A, Bergami A, Libera DD, Benussi L, et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014; 21: 582-593.
- Bianco F, Pravettoni E, Colombo A, Schenk U, Möller T, Matteoli M, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol. 2005; 174: 7268-7277.
- Lalli MA, Bettcher BM, Arcila ML, Garcia M, Guzman C, Madrigal L, et al. Whole-genome sequencing suggests a chemokine gene cluster that modifies age at onset in familial Alzheimer's disease. Mol Psych. 2015.
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011; 14: 724-738.
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994; 91: 10625-10629.
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996; 16: 675-686.
- Goldman SA, Nedergaard M, Windrem MS. Modeling cognition and disease using human glial chimeric mice. Glia. 2015; 63: 1483-1493.
- Maki RA, Tyurin VA, Lyon RC, Hamilton RL, DeKosky ST, Kagan VE, et al. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J Biol Chem. 2009; 284: 3158-3169.
- Kato-Kataoka A, Sakai M, Ebina R, Nonaka C, Asano T, Miyamori T. Soybean-derived phosphatidylserine improves memory function of the elderly Japanese subjects with memory complaints. J Clin Biochem Nutr. 2010; 47: 246-255.
- Zanotta D, Puricelli S, Bonoldi G. Cognitive effects of a dietary supplement made from extract of Bacopa monnieri, astaxanthin, phosphatidylserine, and vitamin E in subjects with mild cognitive impairment: a noncomparative, exploratory clinical study. Neuropsychiatr Dis Treat. 2014; 10: 225-230.
- Wang H, Eckel RH. What are lipoproteins doing in the brain? Trends Endocrinol Metab. 2014; 25: 8-14.
- Wang H, Eckel RH. What are lipoproteins doing in the brain? Trends Endocrinol Metab. 2014; 25: 8-14.
- Reed B, Villeneuve S, Mack W, DeCarli C, Chui HC, Jagust W. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 2014; 71: 195-200.
- Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, et al. Unique lipoproteins secreted by primary astrocytes from wild type, apoE (-/-), and human apoE transgenic mice. J Biol Chem. 1999; 274: 30001-30007.
- Demeester N, Castro G, Desrumaux C, De Geitere C, Fruchart JC, Santens P, et al. Characterization and functional studies of lipoproteins, lipid transfer proteins, and lecithin: cholesterol acyltransferase in CSF of normal individuals and patients with Alzheimer's disease. J Lipid Res. 2000; 41: 963-974.
- Boulay AC, Mazeraud A, Cisternino S, Saubaméa B, Mailly P, Jourdren L, et al. Immune quiescence of the brain is set by astroglial connexin 43. J Neurosci. 2015; 35: 4427-4439.
- Lee M, McGeer E, McGeer PL. Neurotoxins released from interferon-gamma-stimulated human astrocytes. Neuroscience. 2013; 229: 164-175.
- Meraz-Ríos MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernández J, Campos-Peña V. Inflammatory process in Alzheimer's Disease. Front Integr Neurosci. 2013; 7: 59.
- Sailasuta N, Harris K, Tran T, Ross B. Minimally invasive biomarker confirms glial activation present in Alzheimer's disease: a preliminary study. Neuropsychiatr Dis Treat. 2011; 7: 495-499.
- Rodriguez-Vieitez E, Ni R, Gulyas B, Toth M, Haggkvist J, Halldin C, et al. Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: a correlative positron emission tomography and in vitro imaging study. Eur J Nucl Med Mol Imaging. 2015; 42: 1119-1132.
- Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013; 61: 1113-1121.
- Sciacca FL, Ferri C, Licastro F, Veglia F, Biunno I, Gavazzi A, et al. Interleukin-1B polymorphism is associated with age at onset of Alzheimer's disease. Neurobiol Aging. 2003; 24: 927-931.
- Liu L, Chan C. IPAF inflammasome is involved in interleukin-1β production from astrocytes, induced by palmitate; implications for Alzheimer's Disease. Neurobiol Aging. 2014; 35: 309-321.
- Liu L, Chan C. The role of inflammasome in Alzheimer's disease. Ageing Res Rev. 2014; 15: 6-15.
- Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003; 971: 197-209.
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003; 9: 453-457.
- Jones RS, Minogue AM, Connor TJ, Lynch MA. Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J Neuroimmune Pharmacol. 2013; 8: 301-311.
- Gasic-Milenkovic J, Dukic-Stefanovic S, Deuther-Conrad W, Gartner U, Munch G. Beta-amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon-gamma and 'advanced glycation endproducts' in a murine microglia cell line. Eur J Neurosci. 2003; 17: 813-821.
- Norris PC, Gosselin D, Reichart D, Glass CK, Dennis EA. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc Natl Acad Sci U S A. 2014; 111: 12746-12751.
- Hoozemans JJ, Rozemuller AJ, Veerhuis R, Eikelenboom P. Immunological aspects of alzheimer's disease: therapeutic implications. BioDrugs. 2001; 15: 325-337.
- Yermakova AV, O'Banion MK. Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer's disease. Neurobiol Aging. 2001; 22: 823-836.
- Bazan NG, Lukiw WJ. Cyclooxygenase-2 and presenilin-1 gene expression induced by interleukin-1beta and amyloid beta 42 peptide is potentiated by hypoxia in primary human neural cells. J Biol Chem. 2002; 277: 30359-30367.
- Hjorth E, Zhu M, Toro VC, Vedin I, Palmblad J, Cederholm T, et al. Omega-3 fatty acids enhance phagocytosis of Alzheimer's disease-related amyloid-β42 by human microglia and decrease inflammatory markers. J Alzheimers Dis. 2013; 35: 697-713.
- Fattahi MJ, Mirshafiey A. Positive and negative effects of prostaglandins in Alzheimer's disease. Psychiatry Clin Neurosci. 2014; 68: 50-60.
- Johansson JU, Woodling NS, Wang Q, Panchal M, Liang X, Trueba-Saiz A, et al. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer's disease models. J Clin Invest. 2015; 125: 350-364.
- Ong WY, Farooqui T, Kokotos G, Farooqui AA. Synthetic and natural inhibitors of phospholipases A2: their importance for understanding and treatment of neurological disorders. ACS Chem Neurosci. 2015; 6: 814-831.
- Cudaback E, Jorstad NL, Yang Y, Montine TJ, Keene CD. Therapeutic implications of the prostaglandin pathway in Alzheimer's disease. Biochem Pharmacol. 2014; 88: 565-572.
- Dalkara T, Alarcon-Martinez L. Cerebral microvascular pericytes and neurogliovascular signaling in health and disease. Brain Res. 2015; .
- Busshoff U, Hein A, Iglesias A, Dörries R, Régnier-Vigouroux A. CD1 expression is differentially regulated by microglia, macrophages and T cells in the central nervous system upon inflammation and demyelination. J Neuroimmunol. 2001; 113: 220-230.
- Popa N, Cedile O, Pollet-Villard X, Bagnis C, Durbec P, Boucraut J. RAE-1 is expressed in the adult subventricular zone and controls cell proliferation of neurospheres. Glia. 2011; 59: 35-44.
- Vitali C, Wellington CL, Calabresi L. HDL and cholesterol handling in the brain. Cardiovasc Res. 2014; 103: 405-413.
- Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015; 16: 358-372.
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science. 2000; 290: 1768-1771.
- Blazar B R, Lindberg FP, Ingulli E, Panoskaltsis-Mortari A, Oldenborg PA, Iizuka K, et al. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med. 2001; 194: 541-549.
- Oldenborg PA, Gresham HD, Lindberg FP. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J Exp Med. 2001; 193: 855-862.
- Smith RE, Patel V, Seatter SD, Deehan MR, Brown MH, Brooke GP, et al. A novel MyD-1 (SIRP-1alpha) signaling pathway that inhibits LPS-induced TNFalpha production by monocytes. Blood. 2003; 102: 2532-2540.
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987; 262: 14352-14360.
- Morales I, Guzmán-Martínez L, Cerda-Troncoso C, Farías GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer's disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014; 8: 112.
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007; 10: 1538-1543.
- Casano AM, Peri F. Microglia: multitasking specialists of the brain. Dev Cell. 2015; 32: 469-477.
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009; 64: 110-122.
- Neumann H1, Kotter MR, Franklin RJ . Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009; 132: 288-295.
- McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995; 21: 195-218.
- Miller KR, Streit WJ. The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol. 2007; 3: 245-253.
- Jou I, Lee JH, Park SY, Yoon HJ, Joe EH, Park EJ. Gangliosides trigger inflammatory responses via TLR4 in brain glia. Am J Pathol. 2006; 168: 1619-1630.
- Moon DO, Kim KC, Jin CY, Han MH, Park C, Lee KJ, et al. Inhibitory effects of eicosapentaenoic acid on lipopolysaccharide-induced activation in BV2 microglia. Int Immunopharmacol. 2007; 7: 222-229.
- De Smedt-Peyruss V, Sargueil F, Moranis A, Harizi H, Mongrand S, Laye S. Docosahexaenoic acid prevents lipopolysaccharide-induced cytokine production in microglial cells by inhibiting lipopolysaccharide receptor presentation but not its membrane subdomain localization. J Neurochem. 2008; 105: 296-307.
- Blasko I, Stampfer-Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck-Loebenstein B. How chronic inflammation can affect the brain and support the development of Alzheimer's disease in old age: the role of microglia and astrocytes. Aging Cell. 2004; 3: 169-176.
- Jeon SB, Yoon HJ, Park SH, Kim IH, Park EJ. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J Immunol. 2008; 181: 8077-8087.
- Kawashima A, Harada T, Imada K, Yano T, Mizuguchi K. Eicosapentaenoic acid inhibits interleukin-6 production in interleukin-1beta-stimulated C6 glioma cells through peroxisome proliferator-activated receptor-gamma. Prostaglandins Leukot Essent Fatty Acids. 2008; 79: 59-65.
- Wong SW, MKwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009; 284: 27384-27392.
- Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001; 276: 16683-16689.
- Chen YC, Yip PK, Huang YL, Sun Y, Wen LL, Chu YM, et al. Sequence variants of toll like receptor 4 and late-onset Alzheimer's disease. PLoS One. 2012; 7: e50771.
- Dentesano G, Serratosa J, Tusell JM, Ramón P, Valente T, Saura J, et al. CD200R1 and CD200 expression are regulated by PPAR-γ in activated glial cells. Glia. 2014; 62: 982-998.
- Satoi H, Tomimoto H, Ohtani R, Kitano T, Kondo T, Watanabe M, et al. Astroglial expression of ceramide in Alzheimer's disease brains: a role during neuronal apoptosis. Neuroscience. 2005; 130: 657-666.
- Corrigan FM, Horrobin DF, Skinner ER, Besson JA and Cooper MB. Abnormal content of n-6 and n-3 long-chain unsaturated fatty acids in the phosphoglycerides and cholesterol esters of parahippocampal cortex from Alzheimer's disease patients and its relationship to acetyl CoA content. Int J Biochem Cell Bio. 1998; l30: 197-207.
- Zhang H, Ma Q, Zhang YW, Xu H. Proteolytic processing of Alzheimer's β-amyloid precursor protein. J Neurochem. 2012; 120 Suppl 1: 9-21.
- Yang Y, Keene CD, Peskind ER, Galasko DR, Hu SC, Cudaback E, et al. Cerebrospinal Fluid Particles in Alzheimer Disease and Parkinson Disease. J Neuropathol Exp Neurol. 2015; 74: 672-687.