
Research Article
J Immun Res. 2021; 7(1): 1037.
Characterization of the Oral Microbiome in Canine Chronic Ulcerative Stomatitis
Anderson JG1,2#*, Paster BJ3,4#, Kokaras A5 and Chen T3,4
1Sacramento Veterinary Dental Services, Rancho Cordova, California
2University of Pennsylvania, School of Dental Medicine, Philadelphia, USA
3The Forsyth Institute, Cambridge MA, USA
4Harvard School of Dental Medicine, Boston, MA, USA
5New England Bio Labs, England
#Equally Contributed
*Corresponding author: Jamie G. Anderson, University of Pennsylvania, School of Dental Medicine, Philadelphia, USA
Received: March 16, 2021; Accepted: April 20, 2021; Published: April 27, 2021
Abstract
Canine chronic ulcerative stomatitis is a debilitating, oral mucosal disorder of dogs. A commonly held hypothesis for pathogenesis is that bacterial plaque on tooth surfaces is responsible for the ulcerative mucosal lesions. As such, therapy has focused on full-mouth, tooth extraction. Recent studies revealed a unique leukocyte profile in canine ulcerative stomatitis that is amenable to immune modulating therapy. What remains unknown is the role bacteria may play in dysbiosis and immune-inflammatory mechanisms. The microbiota of canine ulcerative stomatitis has not been characterized. Aims of the present study include determination of the microbiome of mucosal lesions in canine ulcerative stomatitis and that of the supragingival plaque of the opposing tooth. The microbiota of these surfaces was compared to healthy mucosa in the canine ulcerative stomatitis patient, and three non-stomatitis control patients. Our hypothesis was that specific microbial species or complexes are associated with ulcerative stomatitis. DNA from 100 clinical samples was evaluated using Next Generation Sequencing methods and was analyzed using LDA Effect Size and the non-parametric factorial Kruskal-Wallis sum-rank test. Statistically significant differences in species were determined from mucosal ulcers versus normal sites in ulcerative stomatitis patients. Species that were more prevalent on the ulcer lesions included putative periodontal pathogens, such as a Tannerella forsythia-like phylotype and Porphyromonas gingivicanis, a species related to the human pathogen Porphyromonas gingivalis. The microbial profile of the supragingival plaque of the abutting tooth to the ulcer revealed similar pathogens. This study showed that in dogs with stomatitis, the mucosal ulcer is inhabited by a unique, species-specific bacterial community and suggests significant differences between the oral mucosa of healthy dogs, dogs with severe periodontal disease, or dogs with oral mucosal tumors. Based on our results, full-mouth, tooth extraction may not be the optimal treatment of the disease.
Keywords: Canine chronic ulcerative stomatitis; Plaque biofilm; Microbiome
Abbreviations
CCUS: Canine Chronic Ulcerative Stomatitis; PD: Periodontal Disease; PCR: Polymerase Chain Reaction; OTU’s: Oral taxonomic unit’s; COHAT: Comprehensive Oral Health and Assessment; OLP: Oral Lichen Planus
Introduction
The clinical, radiographic, and histologic appearance of Canine Chronic Ulcerative Stomatitis (CCUS), including immunohistochemistry and immunofluorescence has recently been described [1,2]. Clinically, CCUS has a unique presentation of deep mucosal, palatoglossal and lingual ulcers that are opposite teeth with heavy plaque biofilm accumulation. There is a fetid malodor associated with the clinical disease and symptomatic patients are painful. Prominent histologic findings include a dense lichenoid lymphocytic-plasmacytic infiltrate at the interface between the mucosal epithelium and subepithelial connective tissue represented by CD79a B cells, plasma cells, CD3 T cells, Fox P3+ cells, equal numbers of CD4+ and CD8+ cells, large numbers of CD3-/IL-17+ cells, macrophages and mast cells [1]. These initial studies helped to reveal that CCUS is an inflammatory immune mediated disease with distinct differences from normal healthy dogs and those with severe periodontitis. CCUS has been reported not to be strongly influenced by age, sex or Periodontal Disease (PD) status; though the Terrier breed may be over-represented [1]. Interestingly, there is a pattern of leukocyte subsets in CCUS inflammatory lesions, differing markedly between normal controls, severe periodontitis [2] and canine oral neoplasms (unpublished data).
The antigenic triggers in CCUS remain poorly defined. It seems likely that, as in periodontitis, the oral microbial population has a role to play in the significant inflammation in CCUS. A commonlyheld hypothesis for CCUS pathogenesis and its “kissing lesions” is that bacterial supragingival plaque from opposing tooth surfaces directly caused the ulcerative mucosal lesions [3-6]. However, the role of bacterial communities in canine mucosal disease has not been studied.
For many years an understanding of human periodontitis was based, in part, on ligature-induced periodontitis studies in beagle dogs [7-9]. The oral microbiome was always assumed to be similar between humans and dogs, and oral diseases were treated similarly between the two mammalian species [10,11]. The first comprehensive description of the subgingival microbiota of people based on cloning and sequencing technology was provided by Paster in 2001 [12]. Since then, techniques based on next-generation sequencing and advanced bioinformatics tools have helped to unravel the complexity of the subgingival microbiota in periodontal health and disease [13-15]. Using these techniques, comprehensive studies on the canine oral microbiome have revealed major differences from the human oral microbiome in health and in mild periodontal disease [16]. In fact, only 16% of the bacterial taxa found in human subgingival plaque was also present in canine subgingival plaque. In dogs, the diverse oral microbiome revealed traditional Gram-negative species being more abundant in subgingival samples of healthy dogs (Porphyromonas was the most abundant genus along with Moraxella and Bergeyella) with Gram-positive species predominating in mild canine periodontitis (Peptostreptococcus, Actinomyces, and Peptostreptococcaceae taxa) [17]. The microbiome of severe periodontitis in dogs has not yet been evaluated with culture-independent techniques.
A recent paper described the microbiome of different niches in dogs’ mouths [18], in which they found three discrete oral niches; soft tissues of the buccal mucosa and dorsum of the tongue, hard tissue supragingival plaque and saliva. The most abundant taxa differed by location. Core microbiota for the buccal mucosal surfaces included four Oral Taxonomic Units (OTU’s); an unclassified Bergeyella sp., an unclassified Capnocytophaga sp., Porphyromonas cangingivalis and an unclassified Porphyromonas sp. In humans, black-pigmented taxa of the phylum and motile organisms have been found on oral mucosal surfaces with and without periodontitis [19,20] and proportions of bacterial species differed greatly depending on location [21,22]. Other studies have demonstrated that healthy individuals are often colonized with different microbiomes than those with disease involving various organ systems [23]. These discoveries combined with the distinct lack of streptococcal species [16,24] in canines suggests that any extrapolation from the human model to the oral colonization process in dogs may be inaccurate. Additionally, the pH of dog saliva is much more alkaline (pH 8.5) [5] than human saliva making it less favorable for acidogenic streptococci.
Historically, Socransky & Haffajee in 1994 [25] reviewed the evidence to support the hypothesis that periodontal disease is caused by bacteria. Further, the model of bacterial plaque biofilm development conceptualized by Socransky and colleagues detailed that a transition occurred from health to disease, or respectively from typical Gram-positive species to Gram-negative species [26]. Current data suggests that specific plaque-associated microbes do not necessarily lead to periodontitis [27]. The keystone-pathogen hypothesis proposes that certain low-abundance microbial pathogens can orchestrate oral inflammatory disease by transforming a normally benign microbiota into a dysbiotic one in a susceptible host [28]. Recognition of alterations to the structure of complex commensal communities can modulate innate and adaptive immune responses and lead to the development of immune mediated inflammatory disorders arising as a result of increases in pathogenic microbiota and diminished numbers of non-pathogenic species [29,30]. The relevance of these mechanisms in PD to the disease process in CCUS is under investigation.
The pathogenesis of oral cancer in humans is complex. Studies of the microbiome associated with oral cancer have revealed new species and uncovered various differences between healthy persons and patients with oral cancer [31]. The possible role of Porphyromonas gingivalis in the development of orodigestive cancers has received significant attention [32,33]. This study included the microbiome evaluation of canine mucosal tumors to determine if commonality in species existed between the CCUS tissue and that of mucosal oral neoplasms.
This is the first investigation of the microbiome of the condition called CCUS. PCR and/or culture-based analyses for this condition are not found in the current literature. In order to better understand the potential role of specific species or microbial complexes of bacteria in the pathological process of CCUS, we applied cultureindependent next generation sequencing methods. Until 2017, the mechanisms operating in CCUS were characterized as idiopathic. This study details the microbiome in CCUS and provides further insights into its pathogenesis and proposed treatment of dogs with this painful condition. The knowledge gained will enhance our ability to medically treat this condition, rather than extracting functional teeth.
Materials and Methods
Ethical statement
Sampling of plaque biofilm is commonly performed during the Comprehensive Oral Health Assessment and Treatment (COHAT) of periodontal disease in companion animals. Supragingival plaque sampling is a non-invasive procedure where sterile paper points are swiped along a plaque retentive surface. For all of the dogs in this study, plaque sampling was performed under general anesthesia as part of the routine COHAT (induction with Propofol 4 mg/kg and Valium 0.5 mg/kg intravenous, maintenance with isoflurane inhalant), with appropriate regional local anesthesia (Bupivacaine 0.5% ml amount dependent on animal size) and postoperative analgesic (Hydromorphone 0.05 mg/kg subcutaneous) administration. Standard veterinary private practice hospitals, as opposed to Veterinary Medical Teaching Hospitals, do not employ IACUC protocols. As such, we utilized and conformed to the American Animal Hospital Association Guidelines for Dental Care and ethics [34]. Additionally, the Academy for Veterinary Dentistry approved this grant proposal (and the ethics of such) in awarding funding. Client consent forms detailing the study were discussed and signed. No adverse events were documented as a result of plaque acquisition.
Study design
This was a descriptive study of the bacterial microbiome within the mucosal and hard tissues of client-owned dogs that were diagnosed with chronic ulcerative stomatitis, based on prior published clinical criteria and histopathology [1]. Negative control dogs were represented by normal healthy dogs, dogs with severe periodontal disease, and dogs with oral tumors. Randomization was not applicable.
Animals and clinical assessment
Thirty-six dogs with CCUS were prospectively enrolled in this study from the clinical caseloads of the first author (JG Anderson, 24 cases), and other veterinary dentists (M Gates, B Stapelton, S Hoffman, A Stone; total 12 cases). Breed, age and sex predisposition were similar to a cohort of CCUS cases published previously [1]. In a subsequent CCUS study population, no significant differences in leukocyte subsets were found with breed, age, sex or periodontal disease status [2]. Negative control samples were obtained from 25 non-CCUS dogs; three healthy dogs, eight dogs with oral mucosal tumors, and 14 dogs with severe periodontal disease, >75% attachment loss.
The negative control cases were presented for dental procedures; either routine dental cleanings, treatment of severe periodontal disease, or assessment of oral neoplasms.
Unfortunately, breed, age and sex matched controls were not represented. Samples were not pooled. The animal sampling data set is presented in “Supplement 1-S1 Table”.
Patient guardians enrolled by all clinicians read and signed a patient release form. “Supplement 2-S2 Table”. Animals were housed in their home environment. Inclusion criteria included two or more chronic erosions or ulcers in the buccal mucosal tissue opposite teeth. Exclusion criteria included animals in renal failure, those with previously diagnosed autoimmune or immune-mediated disease and those dogs that were currently receiving or had received antibiotics or immune-suppressing drug therapy in the past 4 weeks.
Sample collection
Bacterial microbiome samples were collected in the same manner from all sites. Prior to oral irrigation and any tissue manipulation, and in an aseptic fashion, 3 to 5 sterile endodontic paper points were gently swiped across the sample site surface, and then placed into separate sterile plastic tubes, labeled and frozen at -80ºC. Samples were shipped in bulk on dry ice to the Forsyth Institute for DNA isolation and subsequent 16S rRNA gene sequencing. Samples were stored at -80ºC until use.
DNA isolation
Samples were thawed and cells were lysed using a modified protocol using Ready-LyseTM (Lucigen, Middleton, WI) for overnight incubation and subsequently using MasterPure DNA Kit (Lucigen) as described by the manufacturer.
Sequencing
A modification of the protocol as described by Caporaso et al. [35] was used for 16S rDNA sequencing. Briefly, 10-50 ng of isolated DNA was PCR-amplified using V3-V4 primers and 5 PrimeHotMaster Mix (QuantaBio, Beverly, MA). PCR samples were purified using AMPure beads (Beckman Coulter Life Sciences, Indianapolis, IN). 100 ng of each library was pooled, gel-purified, and quantified using a bioanalyser and with qPCR. 12 pM of the library mixture spiked with 20% Phix, was run on a MiSeq (Illumina, San Diego, CA). Negative controls with no added DNA were used as controls.
Sequence de-noising, DADA2 program
16S sequences were processed further using a custom in-house pipeline that takes advantage of both the advanced amplicon denoising algorithm of the recently available DADA2 program [35] and the extensive collection of 16S sequences in the Human Oral Microbiome Database (HOMD) [36]. Briefly, pair-end reads were quality-filtered, merged and clustered using the DADA2 package to obtain OTU tables representing all unique sequences with PCR and sequencing errors accounted for by an error probability model designed by DADA2. An average about 60,000 sequences of about 441 bp per sequence were obtained after bad reads and chimeric sequences had been removed from the analyses. Taxonomy assignment of the representing sequences up to genus level were obtained using a naïve Bayesian classifier [37] as implemented in the DADA2 package. Further classification of the sequences to species level were achieved by string search using the following curated data bases: HMT RefSeq V15.1: 998 sequences, HOMD RefSeq Extended V1.11: 151 sequences, GreenGeneGold V1: 2,623 sequences; NCBI 16S rRNA Reference: 18,044 sequences. This represents 21,816 total unique sequences of which represents 14,651 total species. Note that sequences of <1000nt have been removed. Specific canine oral taxa (COT) based on comparisons of 16S rRNA gene sequences have been previously described (Dewhirst et al., 2012 [16]). These sequences were deposited in GenBank and were available for analysis in this study. The threshold cutoff for species identification was 98.5% similarity. Raw sequencing data and metadata are available online at the Human Oral Microbiome website: http://www.homd.org/ftp/ publication_data/20190404.
LefSE analysis
Sequences of microbial DNA were analyzed using LDA Effect Size (LEfSe), a 2-stage statistical analysis [38]. Specifically, it uses the non-parametric factorial Kruskal-Wallis (KW) sum-rank test to detect features with significant differential abundance with respect to the class of interest and then biological significance is determined using a set of pairwise tests among subclasses using the (unpaired) Wilcoxon rank-sum test. For the output, LEfSe uses Linear Discriminant Analysis to estimate the effect size of each differentially abundant feature.
Results
Sampling
In total, 59 samples were collected from the 36 CCUS patients. Only the lesional mucosa was sampled in 18 cases, in seven cases all three sites were sampled (lesional mucosa, opposing tooth surface supragingival plaque, normal mucosa positive control), and in nine cases the mucosal lesion and the opposing tooth surface supragingival plaque were sampled.
A total of 41 negative control samples were obtained from three healthy non-CCUS dogs (each case sampled three times (mucosa, tooth surface supragingival plaque and a second mucosal site), eight dogs with oral mucosal tumors (six dogs sampled from the mucosal surface of the tumor and two were sampled in the three locations: tumor mucosa, normal mucosa and supragingival tooth surface), and 14 dogs with severe periodontal disease (11 sampled from the subgingival pocket and, three sampled from mucosa, tooth surface supragingival plaque and subgingival pocket).
The oral tumors included a papillary squamous cell carcinoma, a solid carcinoma (Pan CK+/Mel A neg), an adenocarcinoma (Pan CK IHC+), a plasmacytoma (Mum 1+, CK and Synaptophysin neg), a spindle cell sarcoma, a melanoma (Melan A+), a canine acanthomatous ameloblastoma, and a benign peripheral odontogenic fibroma.
Microbiome of CCUS
The insights gained about the microbiome in CCUS mirror our hypothesis that specific microbial species or complexes are associated with the disease. These species are unique to the mucosal lesion in the disease, and not found in normal sites in the CCUS mouth or in healthy controls. One species found in the mucosal microbiota was prevalent in the supragingival plaque of the opposing tooth surface.
Oral microbiome in CCUS mucosal lesions is different as compared to normal control mucosa in CCUS animals
As shown in Figure 1, there were several species that had statistically significant differences between the two groups. Some were known canine species, and many were phylotypes not previously described, likely representing new species (designated as sp. nov. in Figure 1).
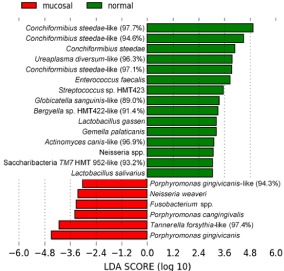
Figure 1: Oral microbiome in CCUS mucosal lesions (class: mucosal)
compared to normal positive control mucosa (class: normal) in CCUS animals.
LefSe analysis illustrating statistically significant differences between the 2
groups. Putative health-associated species are shown in green and CCUS
lesion-associated pathogens are shown in red. An LDA of >2 is considered
statistically significant.
Those species that were more prevalent in CCUS lesions are likely putative periodontal pathogens, i.e., Porphyromonas cangingivalis and P. gingivcanis, 2 canine species related to P. gingivalis, and a T. forsythia-like phylotype. Distribution of these pathogenic species in individual dogs can be readily seen in Figure 2A, 2B, & 2C. Note that the relative abundance of P. gingivicanis (Figure 2B) was very high in CCUS mucosa with an average of 6% of the total population. In Figure 2C, T. forsythia-like phylotype was prevalent on the CCUS lesional mucosa, but not in mucosal plaque from a normal site in the CCUS dog (positive control).
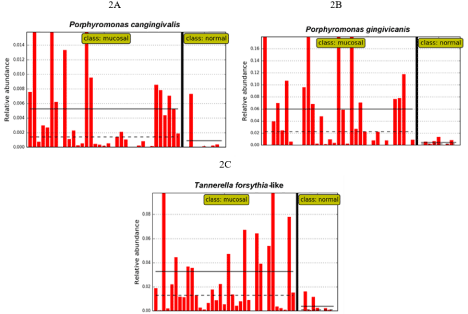
Figure 2: Distribution of select putative pathogens associated with CCUS lesions.
Data from Figure 1. These species are clearly more abundant in CCUS mucosal lesions (class: mucosal) than in mucosal plaque from a normal site in the CCUS dog, positive control site (class: normal). A. P. cangingivalis; B. P. gingivicanis; C. A T. forsythia-like phylotype.
The T. forsythia-like phylotype, present in the CCUS mucosa as above (class: mucosal), was also prevalent in supragingival plaque (class: tooth) in the abutting tooth of CCUS lesions, but not present in the normal CCUS mucosa (class: normal) nor in the supragingival plaque in healthy control dogs (class: healthy) (Figure 3). This indicates that this species may be associated with CCUS pathogenesis and that the supragingival plaque of the abutting tooth may be a potential reservoir.
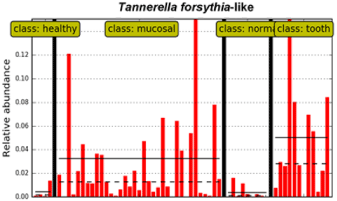
Figure 1: Potential supragingival reservoir of T. forsythia-like phylotype for
CCUS mucosal colonization (class: tooth).
Conversely, the most predominant species in control mucosa in CCUS animals (class: normal) was of the genus Conchiformibius as also indicated in Figure 1. In particular, one phylotype of C. steedae was present at very high relative abundance in most animals from 30 to 50% of the total bacterial population (Figure 4).
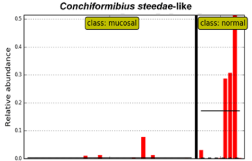
Figure 4: Distribution of species in normal sites in CCUS animals.
Data from Figure 1. High abundance of Conchiformibius can be readily seen in non-infected mucosal sites in CCUS animals (class: normal); and minimal abundance in the lesional mucosa (class: mucosal).
Microbiome of severe periodontitis
When subgingival plaque from severe periodontitis sites was compared to supragingival sites in CCUS animals, 28 species or new phylotypes were more associated with periodontitis including putative periodontal pathogens, e.g., Porphyromonas spp, Fusobacterium spp. and Prevotella spp. (Figure 5). Interestingly, a novel phylotype of TM7, which are known to be parasites of other bacteria [39], was commonly detected. TM7 has also shown relative abundance in chronic periodontitis of humans [40].
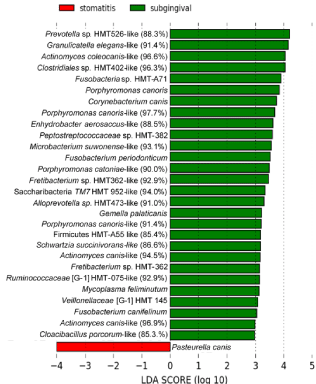
Figure 5: Severe periodontitis associated microbiome (class: subgingival),
and supragingival CCUS (class: stomatitis).
Conversely, only one species, Pasteurella canis, was more associated with supragingival tooth surfaces in CCUS samples. The distribution of P. canis among animals can be clearly seen in Figure 6.
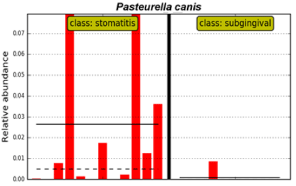
Figure 6: Distribution of P. canis in supragingival plaque in CCUS animals
(class: stomatitis) as compared to subgingival plaque of periodontitis sites
(class: subgingival).
Microbiome of oral neoplasms
Twenty-one novel species were recovered from oral neoplasms sampled in this study. These included five species of Porphyromonas, and numerous other abundant species. This tumor-based population of organisms was statistically different from CCUS lesional mucosa and normal mucosa.
Oral microbiome in CCUS mucosal lesion compared to tumor mucosal surface
Statistically significant differences were seen when comparing CCUS lesions vs surfaces of tumors as shown in Figure 7. The dogs with tumors did not have CCUS.
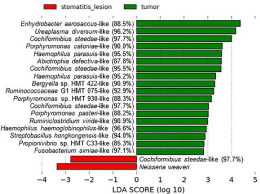
Figure 7: Oral microbiome in CCUS mucosal lesion (class: stomatitis lesion)
compared to tumor mucosal surface (class: tumor). LefSe analysis showing
statistically significant differences between the 2 groups.
In comparing tumor surfaces to noninfected sites in CCUS animals, marked differences were observed (Figure 8), including prevalence of three species of Porphyromonas in the tumor sites. The species associated with non-infected mucosal lesions are likely commensals, e.g., species of Lactobacillus, Streptococcus, Capnocytophaga and Corynebacterium.
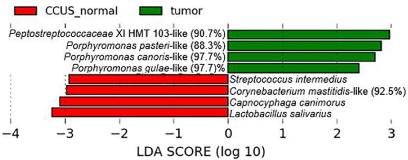
Figure 8: Oral microbiome in noninfected mucosa in CCUS animals (class:
CCUS normal) compared to tumor mucosal surface (class: tumor).
LefSe analysis showing statistically significant differences between the 2 groups
In “Supplement 3-S3 Table” the microbial phylum, genus and species of bacteria identified in CCUS samples, in health, in severe periodontitis, and in oral neoplasms is presented along with the designation of novel species [16].
Discussion
In this study, we characterized the composition of the microbiota in the oral cavity of dogs with and without disease. Our specific goal was to evaluate the oral bacterial microbiome of canine chronic ulcerative stomatitis. We found that the mucosal lesion in this disease is unique and differs from positive normal mucosal controls, healthy mucosal controls and other oral disease conditions such as severe periodontal disease and oral tumors. Numerous phylotypes not previously described were identified and likely represent new species; though their role in disease causation cannot yet be confirmed.
The bacterial microbiome of the ulcerative lesion of CCUS was predominated by three species of Porphyromonas, Neisseria weaveri, Fusobacterium spp, and a T. forsythia-like phylotype. For comparison, there are no published studies representing the microbiome of ulcerative conditions in dogs. That these species are likely putative periodontopathogens may be consistent with samples obtained from subgingival sites in severe periodontitis controls, where 28 species were more associated with severe periodontitis with two species of Porphyromonas, and several species of Fusobacterium and Prevotella. Although it is difficult to draw parallels to humans, many of these taxa are known to be associated with human periodontal disease [41].
Supragingival plaque which accumulates on tooth surfaces above the gum line has been described in dogs by Holcombe et al. [42] and Ruparell et al. [18]. In their studies the most common phyla on tooth surfaces revealed members of Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria.
The positive control (CCUS normal mucosa) and negative control (healthy animal mucosa) tissue revealed species that were very similar to normal mucosal sampling in the recent study which profiled and compared the microbiota of different niches within the oral cavity of dogs [18]. They found the buccal mucosa was dominated by members of the phyla Proteobacteria, Bacteroidetes and Firmicutes. In our study, the most predominant taxa in control mucosa in CCUS animals was of the genus Conchiformibius, a member of the Proteobacteria. Conchiformibius steedae was present in most normal animals at a strikingly high relative abundance, i.e., 30 to 50% of the total population.
The microbiome of mild periodontitis in dogs using non culturebased techniques has been described [17], It is interesting to note that most species found in mild periodontitis were not represented in our sampling of periodontal pockets from severe periodontitis, other than members of Firmicutes. Additionally, other species of abundance in canine severe periodontitis included two species of Actinomyces, several species of Porphyromonas, three species of Fusobacterium and a Synergistes phylotype. This suggests that the microbiome of mild periodontitis and severe periodontitis in dogs, utilizing sequencing technology, are different.
Our results suggest that the CCUS lesion is characterized by an altered microbiome in which there is an increased abundance of putative pathogenic species. However, the presence of pathogenic bacteria within the lesion is not sufficient to attribute disease putative etiology separate from immune-inflammatory mechanisms [2] or local environmental changes [43,44].
As mentioned, normal mucosal sites in canines afflicted with CCUS revealed bacterial species that were similar to normal healthy control mucosa including species of Conchiformibius, Ureaplasma diversum, and Lactobacillus gasseri. These data suggest that the ulcerative lesion and associated inflammation may alter the normal microbiome or predispose to dysbiosis.
In people, there are several reports of specific oral microbiomes being associated with oral and distant malignancies, such as esophageal and pancreatic cancer [45-47]. Fusobacterium has been reported to be a proinflammatory pathogen in laryngeal carcinoma [48] and in colorectal cancer [49,50]. Another study has shown that specific species have been identified that correlate strongly with oral cancer, such as Streptococcus sp., Peptostreptococcus sp., Prevotella sp., Fusobacterium sp., Porphyromonas gingivalis, and Capnocytophaga gingivalis [33].
In this study, differences were seen when comparing the microbiomes of stomatitis mucosal lesions and oral tumors of various types. When comparing tumor surfaces to noninfected sites in CCUS animals, marked differences were observed, including prevalence of three species of Porphyromonas in the tumor sites. It is well known that in humans, P. gingivalis, a close relative of these species of Porphyromonas, has numerous virulence factors, including invasins, cytotoxins, proteases, collagenases and P. gingivalis LPS [51] that allow it to be a potent oral pathogen. It will be interesting to determine whether these canine species have similar virulence factors.
Though the tumor cases for comparison were not numerous, statistically significant results were still achieved when comparing tumor surfaces and the stomatitis mucosa. All of the phylotypes from tumor mucosal surfaces likely represented new species inasmuch as the % similarity was well below the cutoff value of 98.5%. However, comparisons of nearly full 16S rDNA sequences of these phylotypes will be necessary to confirm them as new species. To the authors’ knowledge, this is the first description of the microbiome of oral mucosal tumors in dogs. It appears that the microbiome of canine oral cancers differs from their human counterpart. Further study is warranted in dogs to determine the role of the microbiota in tumor pathogenesis.
Our previous publications have suggested parallels between the human condition called Oral Lichen Planus (OLP) and CCUS [1,2]. In OLP, microbiome studies reveal that a periodontal pathogen Treponema denticola damages the epithelial physical barriers, and that Capnocytophaga gingivalis has a trypsin-like protease that likely degrades junctional proteins. Mechanistically, the bacterial invasion of mucosal tissues observed in OLP may be associated with the changes in the mucosal microbiota. Additionally, F. nucleatum, Eikenella corrodens, and T. denticola, which can invade oral epithelial cells [52, 53], were increased in the OLP mucosa. With the exception of Fusobacterium spp., and though definitive conclusions cannot be made, it appears that CCUS mucosal lesion microbiota was dissimilar to OLP. Further investigations are needed to determine how the pathogenic species are functioning in the mucosa of CCUS.
Limitations in the current study include the small number of normal control samples, the lack of breed, age and sex matched controls, and the omission of sampling periodontal pockets in CCUS patients. Nevertheless, valuable information was obtained to define the microbiome of animals with CCUS and to begin to understand the pathogenesis of the disease.
Conclusion
A highly species-rich bacterial community was shown to inhabit the canine oral mucosa. Of central importance, this study demonstrated that CCUS lesions are characterized by an altered microbiome, in which there is an increased abundance of putative pathogenic bacteria and a decreased abundance of commensal bacteria relative to healthy dogs. The majority of species detected in this study had never been isolated before using conventional culture techniques. Significant differences were detected in the composition of microbiota colonizing the mucosal lesion in CCUS dogs as compared to normal sites within the same dog and compared to normal healthy control mucosa. We further conclude that the bacteria present on the tooth surface opposing CCUS ulcers was likely not the source for the ulcer. Consequently, the present treatment for CCUS of extracting teeth may not be a viable remedy for the disease. Clearly, other more targeted or medical therapies are needed.
Acknowledgement
This study was supported in part by a grant from the Academy of Veterinary Dentistry in 2017.
The first author would like to gratefully acknowledge the assistance of Dr’s. M. Gates, B. Stapleton, K. Ford, S. Hoffman and A. Stone for clinical sample collection.
References
- Anderson JG, Peralta S, Kol A, Kass PH, Murphy B. Clinical and Histopathologic Characterization of Canine Chronic Ulcerative Stomatitis. Vet Pathol. 2017; 54: 511-519.
- Anderson JG, Kol A, Bizikova P, Stapelton BP, Ford K, Villarreal A, et al. Immunopathogenesis of canine chronic ulcerative stomatitis. PLoS One. 2020; 15: e0227386.
- Carmichael DT. Diagnosing and treating chronic ulcerative paradental stomatitis. Veterinary Medicine. 2004; 99: 1008-1011.
- Harvey CE, Emily P. Small animal dentistry. St Louis: Mosby. 1993; 413.
- Lobprise HB. Blackwell’s five minute veterinary consult clinical companion: small animal dentistry. 1st edition. Ames, Iowa: Blackwell Pub. 2007; 414.
- Wiggs RB, Lobprise HB. Veterinary dentistry: principles and practice. Philadelphia: Lippincott-Raven Publishers. 1997; 748.
- Renvert S, Wikstrom M, Mugrabi M, Claffey N. Histological and microbiological aspects of ligature-induced periodontitis in beagle dogs. J Clin Periodontol. 1996; 23: 310-319.
- Lindhe J, Hamp SE, Loe H. Experimental periodontitis in the beagle dog. Int Dent J. 1973; 23: 432-437.
- Sorensen WP, Loe H, Ramfjord SP. Periodontal disease in the beagle dog. A cross sectional clinical study. J Periodontal Res. 1980; 15: 380-389.
- Harvey CE. Periodontal disease in dogs. Etiopathogenesis, prevalence, and significance. Vet Clin North Am Small Anim Pract. 1998; 28: 1111-1128.
- Harvey CE. Management of periodontal disease: understanding the options. Vet Clin North Am Small Anim Pract. 2005; 35: 819-836.
- Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001; 183: 3770- 3783.
- Kononen E, Muller HP. Microbiology of aggressive periodontitis. Periodontol 2000. 2014; 65: 46-78.
- Wade WG. Has the use of molecular methods for the characterization of the human oral microbiome changed our understanding of the role of bacteria in the pathogenesis of periodontal disease? J Clin Periodontol. 2011; 38: 7-16.
- Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, et al. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 2008; 87: 1016-1020.
- Dewhirst FE, Klein EA, Thompson EC, Blanton JM, Chen T, Milella L, et al. The canine oral microbiome. PLoS One. 2012; 7: e36067.
- Davis IJ, Wallis C, Deusch O, Colyer A, Milella L, Loman N, et al. A crosssectional survey of bacterial species in plaque from client owned dogs with healthy gingiva, gingivitis or mild periodontitis. PLoS One. 2013; 8: e83158.
- Ruparell A, Inui T, Staunton R, Wallis C, Deusch O, Holcombe LJ. The canine oral microbiome: variation in bacterial populations across different niches. BMC Microbiol. 2020; 20: 42.
- Van der Velden U, Van Winkelhoff AJ, Abbas F, De Graaff J. The habitat of periodontopathic micro-organisms. J Clin Periodontol. 1986; 13: 243-248.
- Van Winkelhoff AJ, Van der Velden U, Winkel EG, de Graaff J. Blackpigmented Bacteroides and motile organisms on oral mucosal surfaces in individuals with and without periodontal breakdown. J Periodontal Res. 1986; 21: 434-439.
- Mager DL, Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Distribution of selected bacterial species on intraoral surfaces. J Clin Periodontol. 2003; 30: 644-654.
- Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000. 2006; 42: 80-87.
- Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. 2017; 17: 219-232.
- Davis EM. Gene Sequence Analyses of the Healthy Oral Microbiome in Humans and Companion Animals. J Vet Dent. 2016; 33: 97-107.
- Socransky SS, Haffajee AD. Evidence of bacterial etiology: a historical perspective. Periodontol 2000. 1994; 5: 7-25.
- Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998; 25: 134-144.
- Teles R, Teles F, Frias-Lopez J, Paster B, Haffajee A. Lessons learned and unlearned in periodontal microbiology. Periodontol 2000. 2013; 62: 95-162.
- Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012; 10: 717-725.
- Hijazi K, Lowe T, Meharg C, Berry SH, Foley J, Hold GL. Mucosal microbiome in patients with recurrent aphthous stomatitis. J Dent Res. 2015; 94: 87S-94S.
- Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014; 16: 1024-1033.
- Shillitoe EJ. The Microbiome of Oral Cancer. Crit Rev Oncog. 2018; 23: 153- 160.
- Olsen I, Yilmaz O. Possible role of Porphyromonas gingivalis in orodigestive cancers. J Oral Microbiol. 2019; 11: 1563410.
- Karpinski TM. Role of Oral Microbiota in Cancer Development. Microorganisms. 2019; 7: 20.
- Bellows J, Berg ML, Dennis S, Harvey R, Lobprise HB, Snyder CJ, et al. 2019 AAHA Dental Care Guidlines for Dogs and Cats. 2019.
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011; 108: 4516-4522.
- Chen T, Yu WH, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford). 2010; 2010: baq013.
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007; 73: 5261-5267.
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011; 12: R60.
- Bor B, Bedree JK, Shi W, McLean JS, He X. Saccharibacteria (TM7) in the Human Oral Microbiome. J Dent Res. 2019; 98: 500-509.
- Ouverney CC, Armitage GC, Relman DA. Single-cell enumeration of an uncultivated TM7 subgroup in the human subgingival crevice. Appl Environ Microbiol. 2003; 69: 6294-6298.
- Krishnan K, Chen T, Paster BJ. A practical guide to the oral microbiome and its relation to health and disease. Oral Dis. 2017; 23: 276-286.
- Holcombe LJ, Patel N, Colyer A, Deusch O, O’Flynn C, Harris S. Early canine plaque biofilms: characterization of key bacterial interactions involved in initial colonization of enamel. PLoS One. 2014; 9: e113744.
- Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. 1994; 8: 263-271.
- Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. 2003; 149: 279-294.
- Hayes RB, Ahn J, Fan X, Peters BA, Ma Y, Yang L, et al. Association of Oral Microbiome With Risk for Incident Head and Neck Squamous Cell Cancer. JAMA Oncol. 2018; 4: 358-365.
- Peters BA, Wu J, Pei Z, Yang L, Purdue MP, Freedman ND, et al. Oral Microbiome Composition Reflects Prospective Risk for Esophageal Cancers. Cancer Res. 2017; 77: 6777-6787.
- Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM, et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2018; 67: 120-127.
- Gong H, Shi Y, Zhou X, Wu C, Cao P, Xu C, et al. Microbiota in the Throat and Risk Factors for Laryngeal Carcinoma. Appl Environ Microbiol. 2014; 80: 7356-7363.
- Shang FM, Liu HL. Fusobacterium nucleatum and colorectal cancer: A review. World J Gastrointest Oncol. 2018; 10: 71-81.
- Brennan CA, Garrett WS. Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat Rev Microbiol. 2019; 17: 156-166.
- Jia L, Han N, Du J, Guo L, Luo Z, Liu Y. Pathogenesis of Important Virulence Factors of Porphyromonas gingivalis via Toll-Like Receptors. Front Cell Infect Microbiol. 2019; 9: 262.
- Rudney JD, Chen R, Zhang G. Streptococci dominate the diverse flora within buccal cells. J Dent Res. 2005; 84: 1165-1171.
- Ji S, Shin JE, Kim YC, Choi Y. Intracellular degradation of Fusobacterium nucleatum in human gingival epithelial cells. Mol Cells. 2010; 30: 519-526.