
Special Issue - Hepatitis B
Austin J Infect Dis. 2021; 8(1): 1045.
Molecular Analysis of Hepatitis B Virus in Mothers-Children Pairs
Chatzidaki V1, Perdikogianni C1*, Paraskevis D2, Iliopoulos I3, Sourvinos G4, Kouroumalis E5 and Galanakis E1
1Department of Paediatrics, Medical School, University of Crete, Heraklion, Greece
2Department of Hygiene and Epidemiology, National and Kapodistrian University of Athens, Greece
3Laboratory of Bioinformatics, Department of Basic Sciences, Medical School University of Crete, Heraklion, Greece
4Laboratory of Clinical Virology, Medical School, University of Crete, Heraklion, Greece
5Department of Gastroenterology, Medical School, University of Crete, Heraklion, Greece
*Corresponding author: Perdikogianni C, Department of Paediatrics, University of Crete, 70013 Heraklion, Greece
Received: March 29, 2021; Accepted: April 24, 2021; Published: May 01, 2021
Abstract
Background: Vertical transmission of Hepatitis B Virus (HBV) is the primary infection source for infants, but little is known on the proportion of children that have acquired HBV from their mothers.
Objective: We investigated the relationship of HBV sequencing in HBVpositive children and their mothers and explored the HBV phylogenetic tree.
Methods: Serum-extracted HBV-DNA from 38 individuals (13 children paired to nine mothers, 16 unpaired infected children) was amplified by polymerase chain reaction and the target region HBV surface glycoprotein (amino acids 40-171) was directly sequenced. Following editing and alignment of these sequences, phylogenetic tree analysis was performed using the neighbourjoining and maximum-likelihood methods.
Results: Analysis was successfully performed in 29 subjects (23 children and six mothers), including six mother-child pairs. All individuals were infected by genotype D. Subgenotype adw3 prevailed (21, 72.4%), followed by ayw2 (4, 13.8%) and ayw3 (4, 13.8%). Among six mother-child pairs, three had identical and three had different subgenotypes. Phylogenetic analysis revealed that HBV sequences from three children did not cluster with their siblings suggesting a different source of infection.
Conclusion: Our findings suggest that HBV subgenotypes in infected children may not be identical to their mothers’ and point to non-vertical HBV transmission in childhood.
Keywords: Hepatitis B virus; Vertical transmission; Non-vertical transmission; Mothers; Children
Abbreviations
HBV: Hepatitis B Virus; HBsAg: Hepatitis B Surface Antigen; Hepatitis B e Antigen; anti HBe: Hepatitis B e antibody; PCR: Polymerase Chain Reaction
Introduction
Hepatitis B Virus (HBV) remains a considerable health problem despite global immunization strategies, with more than 250 million people having chronic liver infection worldwide [1]. Acquisition of infection in early life is associated with chronicity and mother to child transmission provides a reservoir for chronic carriers and increases the risk for cirrhosis and hepatocellular carcinoma. Vertical transmission of HBV seems to be a principal source of infection for infants, but little is known on the proportion of children that have acquired HBV from their mothers. Previous studies using comparison of HBV DNA sequences and phylogenetic analyses have shown that HBV of chronically infected children originates mainly from their mothers [2,3], but increasing evidence exists for horizontal intrafamiliar transmission [2,4]. The source of infection and other factors including viral genotype and subgenotypes may have an impact on the long-term clinical course of chronic HBV infection [3].
Greece is a country with low to moderate HBV endemicity (HBsAg carriage rates of 0.29-2.6 %) [5]. HBV prevalence is higher among populations with limited access to health care services, i.e., the moving Roma population and immigrants from the Balkans and East European countries [5]. The aim of this study was to explore the phylogenetic tree of pre-S/S HBV gene region in a high-risk moving and immigrant population group in a country of intermediate endemicity and to assess the molecular profile of HBV in HBV positive children and their mothers and hence the molecular closeness of HBV between mothers and children.
Subjects and Methods
Subjects
Samples from 38 individuals were analyzed in the study. Serum samples were obtained from 29 children and adolescents, aged five to 18 years (median age 12.7 years), with suboptimal immunoprophylaxis against HBV, positive for HBV since their early childhood and born to HBsAg positive mothers. Among 19 children with known HBeAg status, 14 were HBeAg positive and five were anti-HBe positive. Sera from nine mothers (13 pairs formed) were collected. None of the subjects had received antiviral treatment or suffered from other concomitant liver or immune disease. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008. Informed consent was obtained from all the mothers and the older children prior to their enrolment and the study was approved by the institutional Ethical Committee, School of Medicine, University of Crete.
Methods
Serological tests: Commercially available enzyme-linked immune assay kits were used according to the manufacturer’s instructions to test serological markers of HBV infection.
Extraction of HBV DNA and determination of HBV subtype and genotype: HBV nucleic acid was extracted from 200μl of serum using a commercially available kit (QIAmp DNA Blood Mini Kit, Qiagen, Hilden, Germany) and were amplified by nested PCR using high fidelity DNA polymerase with proof reading activity and primers derived from the well-conserved areas in the S gene region of the HBV genomes of all eight genotypes (A to H) reported so far [6]. The firstround PCR (94°C for 2 min before the start of cycling: 94°C for 30sec, 55°C for 30sec, and 72°C for 90sec, with an additional 7min in the last cycle) was performed for 35 cycles with primers HB095 (sense, 5'-GAG TCT AGA CTC GTG GTG GAC-3') and HB184 (antisense, mixture of two sequences: 5'-CGA ACC ACT GAA CAA ATG GCA CCG C-3' and 5'-CGC ACC ACT GAA CAA ATT GCA C-3'). The second-round PCR for 25 cycles was carried out under the same conditions as the first-round PCR except for extension for 60 sec with primers HB097 (sense, 5'-GAC TCG TGG TGG ACT TCT CTC-3') and S2-2 (antisense, 5'-GGC ACT AGT AAA CTG AGC CA-3'). The amplification product of the first-round PCR was 461 base pairs (bp) (nucleotides (nt) 244 to 704), and that of the second-round PCR was 437bp (nt 251 to 687). Nucleotide numbers were in accordance with a genotype C HBV isolate of 3,215nt (AB033550). The PCR product was gel-purified and DNA fragments were sequenced with a sequencer ABI PRISMA® 3700 Genetic Analyzer.
Determination of HBV subgenotypes: The nucleotide sequences were translated into amino acid sequences according to the openreading frames of the partial S gene and the HBV subtypes were predicted from the amino acids (aa) at positions 122 (Lys/Arg for d/y determinants), 127 (Pro for w1-2, Thr for w3 and Ile for w4) and 160 (Lys for w and Arg for r). Discrimination between ayw1 and ayw2 was based on positions 134 and 159 (Phe and Ala, for ayw1 and Tyr and Gly for ayw2, respectively) [7].
HBV Genotypes Determination and Phylogenetic Analysis: HBV genotype was determined by sequence analysis of a 437 bp fragment from the HBV S gene fragment (primer sequences at both ends were excluded), using the genotyping tool available at the National Library of Medicine’s National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/projects/genotyping/ formpage.cgi) [8]. Sequence alignment was performed using Clustal W [9]. Phylogenetic analysis was carried out using approximatemaximum likelihood (ML) as implemented FastTree using the GTR+cat nucleotide substitution model. Analysis was done on the partial S gene sequences from 35 sequences obtained from the study subjects and all genotype D (N=2,965) sequences available on public database. Statistical robustness of the tree was assessed using the Shimodaira-Hasegawa (SH) values as implemented in FastTree. Phylogenetic trees were visualized using FigTree program. Moreover, phylogenetic analyses were performed on a set of sequences from the study population and the most similar ones identified by BLAST search (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Results
HBV genotypes and subgenotypes in the total cohort
All children and all mothers carried the HBV D genotype (Table 1). Subgenotype was identified in 29 subjects (23 children and six mothers). The analysis showed that adw3 was the most prevalent subgenotype detected in 21 out of 29 subjects (72.4%), followed by ayw2, detected in 4 (13.8%) and ayw3 (4, 13.8%). Direct sequencing was successfully performed in all 29 PCR-amplified samples and the phylogenetic tree is shown in Figure 1.
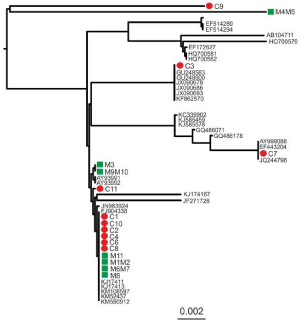
Figure 1: Phylogenetic tree showing sequences from children (red dots) and
their mothers (green boxes) as well as the most closely related identified by
BLAST search. Identification numbers of individuals are reported with “C” and
“M” for children and their mothers, respectively. The accession numbers of
references and the scale of branch length are shown on the tree.
![]()
Age (yrs)
Child
Mother
HBeAg status
HBV genotype /subgenotype
HBeAg status
HBV genotype /subgenotype
1
18
HBeAg (+)
D / adw3
HBeAg (+)
D / adw3
2
11
HBeAg (+)
D / ayw2
HBeAg (+)
D / adw3
3
16
HBeAg (+)
D / ayw3
AntiHBe (+)
D / adw3
4
6
AntiHBe (+)
D / adw3
N/A
D / adw3
5
5
HBeAg (+)
D / ayw3
N/A
D / adw3
6
6
HBeAg (+)
D / adw3
N/A
D / adw3
HBV: Hepatitis B Virus; yrs: Years; HBeAg: Hepatitis B e Antigen; AntiHBe: Hepatitis B e antibody; N/A: Non Applicable.
Table 1: HBV genotype and serotype in six child-mother pairs.
Mother and child pairs
Among six mother-child pairs with successful HBV sequencing, three pairs had identical subgenotype adw3, while three had different subgenotypes; in all three latter cases, children had subgenotype adw3 but mothers had ayw3 (n=2) or ayw2 (n=1). In two further cases, the two siblings born to the same mother did not have the same subgenotype. In each case, one of the siblings had subgenotype identical to their mother’s and the other sibling another subgenotype.
Phylogenetic analysis of HBV sequences is depicted in Figure 1. Identification numbers of individuals are reported with “C” and “M” for children and their mothers, respectively. Analysis of HBV sequences from child sequences and their siblings plus a large dataset of all available genotype D sequences or the most similar sequences identified by BLAST search, revealed that three child sequences (C3, C7 and C9) clustered separately from their siblings (Figure 1), suggesting a different source of infection for these children. Similarly, sequence from M4M5 clustered separately from child C4. On the other hand, sequences from C1, C2, C4, C6, C8, C10, C11 clustered together with M1M2, M3, M6M7, M8 and M9M10 and M11 suggesting that for these cases vertical or intrafamiliar HBV transmission for the pairs C1/M1M2, C2/M1M2, C6/M6M7, C8/M8, C10/M9M10 and C11/M11 cannot be excluded (Figure 1).
Discussion
The findings of this study point to a quite high frequency of subgenotype adw3 in the study area and to the lack of identity between the strains isolated from mother and child in half of the cases. Similarly, to other studies, we chose to focus on a conserved part of the S gene [3] and for the determination of HBV genotype a conserved part of the S gene was repeatedly used. HBV genotype D prevailed in our study population and this is in line with previous reports from the Mediterranean countries and the few existing reports from Greece [9- 12]. Genotypes B and C are prevalent in endemic countries of Asia, where perinatal transmission is the main route of infection [13,14].
Our finding of adw3 predominance differs from previous serotyping results in Greek adults, who carried mostly ayw2 and ayw3 serotypes [11], probably due to the different moving and immigrant population groups in our study. Hadziyannis and LeBouvier conducted a large study of HBsAg subtypes in acute and chronic infection in Greece and found a higher than 90% predominance of ayw2 and ayw3 [12]. Strains encoding adw have so far been described within the A-C and F genotypes [7]. Adw3 as a subtype of D genotype was previously described in individuals with origin from Spain and Sweden [5].
The mode of vertical or horizontal transmission of HBV in early life seems to differ from population to population [12,15]. Previous studies have demonstrated that HBV of chronically infected children originated mainly from their mothers or fathers [2]. The molecular closeness of HBV between adult patients with presumed vertically acquired HBV and their mothers is a strong evidence of vertical transmission even if assessed years after the primary infection [3]. On the other hand, Shen et al found that in a child-mother pair, although the child was infected from the mother (genotype C, subgenotype C2 and serotype adrq), the divergence between them was 0-0.8% and the dominant mutants in the child were different from the maternal [16]. In another study, many mutations emerged in the HBV genome of infants perinatally infected who eventually died from fulminant hepatitis [17].
Our study has several limitations. The cohort was small, but not many children are now a days infected, following perinatal prevention of vertical transmission of HBV. In addition, sequencing was not successfully performed in all the children and mothers of the study, based mainly on the size of specific sequences. Moreover, the size of obtained sequences was not very long, however based on available data, the fact that sequences from at least three children did not cluster with their siblings cannot be doubted. Analysis of larger genomic regions may uncover additional non-linked pairs, but for the purpose of current analysis, we were able to identify at least three unlinked cases.
Conclusion
In conclusion and despite the above-mentioned limitations, our data suggest that besides vertical mother-to-child transmission, other sources may also contribute to acquisition of infection. As the route of transmission correlates with progression of the disease and treatment effectiveness, identification of the source of infection may have considerable clinical impact. Sequencing of viral strains from all family members and the molecular comparison of HBV genotypes and subgenotypes between members of the same family may prove useful in the understanding of the source of chronic HBV infection in childhood.
Acknowledgment
The study was partially funded by the University of Crete Research Account (Grant #3508).
Conflict of Interest
Virginia Chatzidaki, Chrysoula Perdikogianni, Dimitrios Paraskevis, Ioannis Iliopoulos, George Sourvinos, Elias Kouroumalis and Emmanouil Galanakis declare that they have no conflict of interest.
Authors Contributions
VC did the sampling, conducted part of the experimental work and wrote the manuscript, CP analyzed part of the data and wrote the manuscript, DP analyzed the data and reviewed the manuscript, II analyzed the data and reviewed the manuscript, GS conducted part of the experimental work and reviewed the manuscript, EK reviewed the manuscript, EG conceived the idea and reviewed the manuscript, all authors read and approved the final version.
References
- World Health Organization Fact Sheet Hepatitis B. 2020.
- Komatsu H, Inui A, Sogo T, Hiejima E, Kudo N, Fujisawa T. Source of transmission in children with chronic hepatitis B infection after implementation of a strategy for prevention in those at high risk. Hepatol Res. 2009; 39: 569- 576.
- Kim HS, Choi BY, Choi HS, Shin WG, Kim KH, Lee JH, et al. Phylogenetic analyses of HBV Pre S/S genes in mother-child pairs with long –term infection by presumed vertical transmission. J Korean Med Sci. 2014; 29: 564-569.
- Datta S. An overview of molecular epidemiology of hepatitis B virus (HBV) in India. Virology J. 2008; 5: 156-167.
- Zacharakis G, Koskinas J, Kotsiou E, Pouliou E, Papoutselis M, Tzara F, et al. Natural history of chronic hepatitis B virus infection in children of different ethnic origins: a cohort study with up to 12 years’ follow-up in northern Greece. J Ped Gastroenterol Nutr. 2007; 44: 84-91.
- Davaalkham D, Ojima T, Uehara R, Watanabe M, Oki I, Endo K, et al. Analysis of hepatitis B surface antigen mutations in Mongolia: molecular epidemiology and implications for mass vaccination. Arch Virol. 2007; 152: 575-584.
- Magnius LO, Norder H. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology. 1995; 8: 24-34.
- Rozanov M, Plikat U, Chappey C, Kochergin A, Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004; 32: 654- 659.
- Katsoulidou A, Paraskevis D, Magiorkinis E, Moschidis Z, Haida C, Hatzitheodorou E, et al. Molecular Characterization of occult hepatitis B cases in Greek blood donors. J Med Virol. 2009; 81: 815-825.
- Raptopoulou M, Papatheodoridis G, Antoniou A, Ketikoglou J, Tzourmakliotis D, Vasiliadis T, et al. Epidemiology, course and disease burden of chronic hepatitis B virus infection. HEPNET study for chronic hepatitis B: A multicentre Greek study. J Viral Hepat. 2009; 16: 195-202.
- Fylaktou A, Papaventsis D, Daoudaki M, Moskophidis M, Reiberger T, Malisiovas N. Molecular epidemiology of chronic hepatitis B virus infection in Greece. J Med Virol. 2011; 83: 245-252.
- Hadziyannis SJ. Natural history of chronic hepatitis B in Euro-Mediterranean and African countries. Hepatology. 2011; 55: 183-191.
- McMahon BJ. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatology. 2009; 3: 334-342.
- Kang HS, Kang KS, Song BC. Precore and core promoter mutations of the hepatitis B virus gene in chronic genotype C-infected children. J Korean Med Sci. 2011; 26: 546-550.
- Datta S, Banerjee A, Chandra PK, Chowdhury A, Chakravarty R. Genotype, phylogenetic analysis and transmission pattern of occult Hepatitis B Virus (HBV) infection in families of asymptomatic HBsAg carriers. J Med Virol. 2006; 78: 53-59.
- Shen T, Yan XM, Zou YL, Gao JM, Dong H. Virologic characteristics of hepatitis B virus in patients infected via maternal-fetal transmission. World J Gastroenterol. 2008; 14: 5674-5682.
- Friedt M, Gerner P, Wintermeyer P, Wirth S. Complete hepatitis B virus genome analysis in HBsAg positive mothers and their infants with fulminant hepatitis B. BMC Gastroenterol. 2004; 4: 11.