
Research Article
Austin J Infect Dis. 2021; 8(2): 1050.
Are the SARS-CoV-2 Variants Greater Threats? - An In Silico Analysis of the Spike Protein
Jerica Wilson, Viktoriia Sokolova, Katherine C Ji and Hai-Feng Ji*
Department of Chemistry, Drexel University, Philadelphia, PA, USA
*Corresponding author: Hai-Feng Ji, Department of Chemistry, Drexel University, Philadelphia, PA, 19104, USA
Received: May 17, 2021; Accepted: June 03, 2021; Published: June 10, 2021
Abstract
March 2020 marked the beginning of a global pandemic caused by SARSCoV- 2. With the development, production and distribution of several vaccines there are hopes to an end in sight. However, with the emergence of several mutated viral strains concerns are mounting as to the effectiveness of the current treatments and preventative measures against the new strains. Herein we analyzed and compared the interaction of the SARS-CoV-2 Spike (S) protein and its variants with human Angiotensin Converting Enzyme (ACE-2) and the binding affinities of several possible S-protein inhibitors with these variants via in silico molecular docking studies. The binding affinities of all the variants to ACE- 2 are less than that of SARS-CoV-2, indicating they are less potent than SARSCoV- 2. The inhibitors, however, showed decreased binding affinity to most of the mutant S-proteins than SARS-CoV-2, indicating it is more difficult to treat COVID using the therapeutic approach targeting the S-protein.
Keywords: SARS-CoV-2; Angiotensin Converting Enzyme; In Silico
Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARSCoV- 2), which causes the Coronavirus Disease (COVID-19), is a beta coronavirus first reported in December 2019 in Wuhan, China [1]. Since then, SARS-CoV-2 has initiated a global pandemic, with over 140 million confirmed cases of COVID-19 and over 3 million reported deaths [2]. Recently, variants of the virus have emerged and their higher infection rate has become a cause for concern with questions as to whether or not the existing vaccines and established treatments will be able to combat the mutations. The Variants of Concern (VOCs) are the United Kingdom B.1.1.7 variant (N501Y mutation), the South African B.1.351 variant (N501Y-K417N-E484T mutation), the Brazilian P.1 variant (N501Y-K417T-E484T mutation) and the Indian B.1.617 variant (L452R-E484Q mutation). These variants all have mutations that affect the SARS-CoV-2 spike protein (S-protein) Receptor Binding Domain (RBD) [3,4]. The S-protein is composed of 2 subunits, S1 and S2. The RBD of the S1 subunit binds with the human Angiotensin-Converting Enzyme (ACE-2) receptor on host cells then the S2 subunit facilitates fusion of the membranes [5,6]. As the established receptor of SARS-CoV-2 the inhibition of binding to ACE-2 is an important therapeutic route [7,8]. As such, we calculated and compared the binding energies of ACE-2 with the S-proteins of SARS-CoV-2 and the variants. We also performed an in silico investigation of potential S-protein-ACE2 inhibitors then docked the most efficient inhibitors with structures of the S-protein variants. This study is a continuation of our earlier reported work on in silico modeling of the SARS-CoV-2 S-protein and 3CL Protease [9,10].
Methods
Molecular docking calculations were performed using AutoDock Vina 1.1.2 [11]. Ligands were prepared using AutoDockTools-1.5.6 [12], Chimera 1.14 [13] and Avogadro [14]. Proteins were mutated in silico using Schrödinger Maestro [15] and prepared using AutoDockTools-1.5.6. Protein-ligand complexes were visualized with PyMOL [16] and ChimeraX-1.2.1 [17]. Protein-protein docking was performed utilizing the HADDOCK webserver [18].
Preparation of receptor and ligands
The crystal structure of the SARS-CoV-2 S protein RBD bound to ACE-2 was retrieved from the Protein Data Bank (PDB ID: 6M0J). The structure was cleaned, removing ACE-2, and prepared through the addition of polar hydrogens and calculation of Gasteiger charges with AutoDockTools. A databank of FDA-approved drugs was retrieved from BindingDB and optimized with the MMFF94 force field in Avogadro for use as ligands in the docking study.
In silico mutation of spike protein
The SARS-CoV-2 S protein RBD was mutated in silico and then prepared in the above-described methods for the receptor before docking occurred. Using Shrödinger Maestro, individual amino acid residues were selected and altered to match the mutations of the concerning S protein variants. Asparagine 501 was mutated to tyrosine for the N501Y variant, for K417T lysine 417 was mutated to threonine, for K417N lysine 417 was mutated to asparagine, glutamic acid was mutated to lysine for E484K, and glutamine for E484Q and for L452R leucine 452 was mutated to arginine.
Molecular docking
To inhibit the S-protein-ACE-2 interaction, the grid box was constructed to cover all possible docking sites. Set to 22.00 Å × 42.00 Å × 22.00 Å along the x, y and z axis, respectively, and centered at x = -21.878, y = 25.205 and z = 5.514 the grid spans the ACE-2 binding region. The entirety of the FDA-approved drug ligand library was docked with the S-protein and the top ten highest binding ligands by docking score (kcal/mol) were selected. The highest binding affinity being the more negative value. The top ten ligands were then docked with the three mutant S-proteins to determine how the mutations would affect the binding affinity.
Visualization with PyMOL and chimera
The protein-ligand complex of the highest binding affinity for the S-protein was analyzed using both PyMOL and Chimera to visualize the ligand conformation and interactions between the protein and ligand. The complex between this same ligand and the mutated proteins were also visualized, regardless of docking score to compare ligand conformation and interactions.
Protein-protein docking
The HADDOCK webserver was utilized to dock the S-proteins and mutated S-proteins with human ACE-2. PDB files of the S-protein-ACE-2 complexes were built with Maestro and uploaded to the server. Results are reported as a HADDOCK score, a weighted sum of energy terms.
Results
Table 1 compiles the protein-protein docking scores between the S-proteins and ACE-2 as well as the S-protein variants and ACE-2. The top ten FDA-approved drugs with the highest binding affinity to the S-protein RBD are depicted in Table 2 along with the affinities of the same drug bound to the RBD variants. The docking scores are arranged in decreasing affinity in regard to the S-protein RBD. Figure 1 depicts the docking of Irinotecan, the highest binding ligand to S-protein, with all five S-protein variants. Figure 2 illustrates the interactions between the S-protein amino acid residues and the bound ligand, Irinotecan.
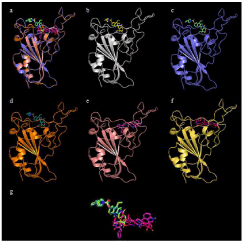
Figure 1: Docking site of Irinotecan with S-proteins RBDs. a) All five
S-proteins bound to irinotecan overlayed; b) Irinotecan bound to S-protein;
c) Irinotecan bound to S-protein N501Y; d) Irinotecan bound to S-protein
N501Y-K417N-E484T; e) Irinotecan bound to S-protein N501Y-K417T-E484T;
f) Irinotecan bound to S-protein L452R-E484Q; g) Overlapping structures of
irinotecan in bound conformation to all 4 proteins.
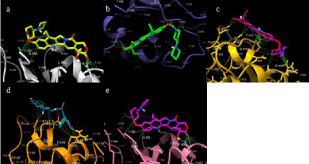
Figure 2: Interactions between Irinotecan and SARS-CoV-2 Proteins. a)
Irinotecan bound to S-protein; b) Irinotecan bound to S-protein N501Y; c)
Irinotecan bound to S-protein N501Y-K417N-E484T; d) Irinotecan bound to
S-protein N501Y-K417T-E484T; e) Irinotecan bound to S-protein L452RE484Q.
![]()
S-protein with ACE-2
HADDOCK Score
SARS-CoV-2 S-protein
-119.9 +/- 2.9
SARS-CoV-2 S-protein N501Y
-109.9 +/- 3.0
SARS-CoV-2 S-protein N501Y-K417N-E484T
-114.5 +/- 4.3
SARS-CoV-2 S-protein N501Y-K417T-E484T
-113.1 +/- 11.4
SARS-CoV-2 S-protein L452R-E484Q
-112.0 +/- 1.2
Table 1: HADDOCK Score of ACE-2 complex with SARS-CoV-2 S-protein or the S-protein variants.
![]()
Drug Name
Docking Score (kcal/mol)
Indication
Original
UK
SA
B
I
Irinotecan
-8.6
-8.2
-6.5
-8.7
-6.2
Metastatic Carcinoma
CAS # 2415492-58-7
-8.5
-8.2
-6.4
-6.5
-7.7
Dihydroergotamine
-8.5
-8.3
-7.4
-7.3
-7.2
Migraine headaches
Lapatinib
-8.4
-7.9
-6.4
-6.3
-5.9
Metastatic Breast Cancer
Nilotinib
-8.4
-8.1
-7.3
-7.3
-7.3
Myeloid Leukemia
Lapatinib ditosylate
-8.4
-8.3
-4.8
-5.2
-6.4
Metastatic Breast Cancer
Digoxin
-8.3
-7.7
-6.9
-6.8
-6.6
Heart Failure
Digitoxin
-8.3
-8.2
-6.8
-6.9
-6.7
Congestive heart failure, atrial fibrillation
CAS # 1610051-53-0
-8.3
-8.4
-6.5
-6.6
-7.1
Ergotamine
-8.3
-8.8
-7
-7.3
-6.8
Migraine headaches
Table 2: Docking Scores of ligands and SARS-CoV-2 S-protein, S-protein N501T (UK), S-protein N501Y-K417N-E484T (SA), S-protein N501Y-K417T-E484T (B), and S-protein L452R-E484Q (I).
Discussion
The binding affinities of all the variants to ACE-2 are less than that of SARS-CoV-2. It is noteworthy that the Brazil variants N501YK417T-E484T showed a larger error range, indicating a higher data variability.
Comparing the ten highest binding affinities of the S-protein and FDA-approved ligands to the binding affinities of variant S-proteins and same ligands shows how the mutations carried by the variants do affect binding at the RBD. The UK variant shares similar docking scores to the S-protein and as such would most likely be susceptible to the same treatment strategies employed against the S-protein. The other three variants, however, greatly deviate from the binding affinities of the S-protein and ligands suggesting these variants may pose a therapeutic issue.
Conclusion
Mutations in the S-proteins receptor binding domain have raised questions as to whether established treatments and the current vaccines will remain effective. This study establishes that inhibitors that would be effective for the S-Protein may prove to be ineffective for three of the S-protein mutations, namely the South African, Brazilian and Indian variants. The UK variant and potential inhibitors appear to share similar binding affinities with the S-protein and potential inhibitors. Three conclusions seem to be drawn via the in silico analysis of the interactions of SARS-CoV-2 and its variants with ACE-2 and the comparison of potential inhibitors for SARSCoV- 2 or these variants. First, the binding affinities of all the variants to ACE-2 are less that of SARS-CoV-2, which is consistent with the general understanding that virus are less potent than their original one after several-generation mutations. Second, even the variants are less potent, most of the inhibitors for the SARS-CoV-2 variants show a significant lower binding affinity to the variants than to SARS-CoV-2, which poses a big challenge to the therapeutic approach targeting the S-protein to treat COVID. Lastly, since the most advanced COVID-19 vaccines, including Pfizer-BioNTech, Moderna, Johnson & Johnson/Janssen, and Oxford/AstraZeneca use cell machinery to make copies of the S-protein, vaccine efficacy and development may also face the similar difficulties towards variants. This suggests that consistent booster shots may be necessary to address future variants. These results emphasize the importance of widespread immunity achieved by current COVID-19 vaccines to prevent a pathway for further mutation of variants, as well as the importance of exploring other methods for targeting COVID-19.
References
- Wu D, Wu T, Liu Q, Yang Z. The SARS-CoV-2 outbreak: What we know. Int J Infect Dis. 2020; 94: 44-48.
- Organization WH. WHO Coronavirus (COVID-19).
- Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature. 2021; 592: 438-443.
- Khan A, Zia T, Suleman M, Khan T, Ali SS, Abbasi AA, et al. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J Cell Physiol. 2021.
- Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. 2020; 117: 11727- 11734.
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020; 181: 281-292.
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020; 581: 215-220.
- Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020; 181: 271-280. e8.
- Hall DC Jr, Ji HF. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med Infect Dis. 2020; 35: 101646.
- Qiao Z, Zhang H, Ji HF, Chen Q. Computational View toward the Inhibition of SARS-CoV-2 Spike Glycoprotein and the 3CL Protease. Computation (Basel). 2020; 8.
- Trott O, AJ Olson. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010; 31: 455-461.
- Sanner MF. Python: A programming language for software integration and development. J Mol Graph Model. 1999; 17: 57-61.
- Eric F Pettersen, Thomas D Goddard, Conrad C Huang, Gregory S Couch, Daniel M Greenblatt, Elaine C Meng, et al. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem. 2004; 25: 1605-1612.
- Marcus D Hanwell, Donald E Curtis, David C Lonie, Tim Vandermeersch, Eva Zurek & Geoffrey R Hutchison. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012; 4.
- Schrödinger L. Schrödinger Release 2021-1: Maestro, New York, NY. 2021.
- Schrödinger L. The PyMOL Molecular Graphics System. Version 2.0. 2015.
- Pettersen EF, GT, Huang CC, Meng EC, Couch GS, Croll TI, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021; 30: 70-82.
- Cyril Dominguez, Rolf Boelens, Alexandre MJJ Bonvin. HADDOCK: a protein-protein docking approach based on biochemical and/or biophysical information. J Am Chem Soc. 2003; 125: 1731-1737.