
Research Article
Austin J Infect Dis. 2022; 9(3): 1072.
Effects of Caspase-3 on HIV-1 Latency in a 3.01 Cells
Wang X*, Huang H, Biswas S, Zhao J, Devadas K and Hewlett I*
Lab of Molecular Virology, Division of Emerging and Transfusion Transmitted Diseases, Center for Biologics Evaluation and Research, USA
*Corresponding author: Xue Wang & Indira Hewlett, Laboratory of Molecular Virology, CBER/FDA, Building 72, Rm 4322, 10903 New Hampshire Avenue, Silver Spring, MD 20993, USA
Received: August 19, 2022; Accepted: September 20, 2022; Published: September 27, 2022
Abstract
Latent infection is a major barrier for cure of HIV-1/AIDS. HIV-1 is capable of establishing latency and the components of the apoptotic pathways may affect viral latency. However, it is not well-known how anti/pro-apoptotic components modulate HIV-1 replication and latency. Using the susceptible A3.01 cell line, we investigated some long-term effects of caspase-3 activities on HIV-1 DNA levels using a sensitive real-time PCR assay. Here we report that viral DNA levels increased upon treatment with caspase-3 inhibitor, Z-DEVD and decreased with caspase-3 activator, PAC1. We also simultaneously measured viral RNA from supernatants of these cell cultures and found that the degree of HIV-1 latency is inversely proportional to levels of viral replication. Furthermore, we demonstrated that inhibition of caspase-3 activities promoted viral latency and inhibited viral replication in several ways, which may include: 1) inhibition of viral RNA un coating with increased Trim5a expression; 2) deleterious mutations in the viral genome with increased APOBEC3G; 3) transcriptional interference with decreased levels of the host factors, NF-κB p65, Ap-1, Sp-1, NFAT, STAT1/3/5, IRF3/7, inactivated YB-1 and MAPK, Erk1/2 and p38, and inhibition of full-length of HIV-1 mRNA and P-TEF b signaling; 4) epigenetic silencing with decreased PCAF; 5) blocking trafficking of the components of viral particle and budding with decreased Tag101 and Alix. These data suggest that HIV-1 infection can employ or even manipulate the cellular apoptotic status to favor viral survival and escape monitoring and destruction by the host immune system.
Keywords: Apoptosis; Caspase-3; HIV-1; Real-time PCR; Latency; Replication
Introduction
Human Immunodeficiency Virus type-1 (HIV-1) infection destroys CD4 T lymphocytes (CD4 cells) of the immune system, leaving the body vulnerable to life-threatening infections and cancers resulting in Acquired Immunodeficiency Syndrome (AIDS). Since the beginning of the epidemic, 76 million people have been infected with the HIV virus and more than 33 million people have died of HIV/AIDS [1]. Antiretroviral Therapy (ART) can keep the virus suppressed and live long and healthy lives. However, the virus continues to persist in long-lived resting CD4+ T cells, macrophages and astrocytes which form a viral reservoir in infected individuals and impede complete eradication of the virus [2]. Clinical data indicate that cessation of ART causes a rapid rebound of viremia in most patients and is accompanied by the emergence of HIV drug resistance mutants, which have been on the increase in recent years [2].Therefore, to maintain control of HIV infection patients may need to periodically modify and change ART regimens as needed.
Apoptotic cell death induced by HIV-1 infection has been reported to be one of the important pathways to cause progressive destruction of CD4+ T cells [3,4]. Accumulating evidence indicates that the accelerated apoptosis of CD4+T cells in HIV infection is multifactorial, with direct viral cytotoxicity and/or indirect killing (bystander killing). HIV-1-induced apoptotic signaling events triggered by viral proteins, such as gp 120, Nef, Tat, Vpu, Vpr and protease, include upregulation of Fas/Fas lig and (FasL), activation of caspase-8 activities and down regulation of FLIP in death receptor (extrinsic)-mediated apoptotic pathways [5], up regulation of Bax, and down regulation of Bcl 2 in mitochondrial (intrinsic)-mediated apoptotic pathways; and increased caspase-3 activation, leading to cell death [3-6].
After infection, HIV-1 manipulates host cells for its reproduction. HIV-1 infection-induced apoptosis results in release of virus from infected cells and virus dissemination in vivo [7]. We had reported that in lymphocytic cells HIV replication (a) increased with expression of proapoptotic proteins such as FasL, FADD, and p53, (b) decreased with expression of anti-apoptotic proteins, Bcl-XL, FLIP, and XIAP, (c) decreased with knockdown of proapoptotic proteins, Bax and FADD, and (d) decrease in inhibition of caspase 3 activity [8-10] and caspas-8 activation was reported to increase HIV-1replication through NF-kB pathway [11,12] suggesting that proapoptotic molecules (such as FasL, FADD, p53, Bax, caspase-3, or caspase-8) promote HIV-1 replication, while antiapoptotic components (such as Bcl2/Bcl-XL, FLIP, or XIAP)from apoptotic pathways inhibit HIV-1 replication.
Cells that carry a latent provirus that is not expressed will not be eliminated by host immune responses [12,13]. To achieve cure of HIV infection, all infectious proviruses need to be eliminated, or replication should be permanently blocked in order to inhibit production of progeny virus [13,14]. However, the mechanisms whereby HIV-1 latent reservoirs may be irreversibly inhibited in vivo and in vitro are unknown at the present time. Recently, we have provided direct evidence that proapoptotic factors, from both death receptor-mediated and mitochondria-mediated apoptotic pathways, decrease HIV-1 latency while antiapoptotic factors increase this state, suggesting that while apoptotic inhibition reduces HIV-1 replication and cytopathogenesis, it also promotes the seeding of latent reservoirs in a manner that enhances virus production when cells are activated [15].
Latently infected cells do not produce virus constitutively but can be induced by T cell activation to produce infectious virus [14]. The reversible lack of viral expression allows survival in longlived CD4+ T cells that can propagate the provirus during cell division [13,14]. Existing literature has indicated that the molecular mechanisms of latency are complex, which may include the lack of nuclear forms of key host transcriptional factors (e.g., NF-κB, NFAT, AP1), epigenetic modifications which hinder HIV-1 gene expression, and the interference of transcriptional efficiency and elongation in host cells [13,14]. Direct exposure of the latently infected promonocytic (U1) and lymphoid (ACH-2) cells to activated caspases could induce viral replication, while pretreatment of the cells with the pan-caspase inhibitor Z-VAD-FMK prior to exposure to the cytotoxic agents inhibited apoptosis and viral activation [16]. HIV-1 virions produced in association with host cell apoptosis were shown to be infectious [15]. Previously, we reported that the proapoptotic molecule, p53, could reactivate HIV-1 replication from its latent state to different levels including upregulation/activation of host transcription factors and recruitment of Histone Acetyl Transferase (HAT)/inhibition of histone deacetylation to acetylate histonetails and to open nucleosomes to facilitate HIV transcription [17].
Here we report that after infection with HIV-1, A3.01 cells treated with caspase-3 inhibitor or activator displayed different sizes of latent reservoirs harboring different viral DNA products. We also investigated mechanisms by which caspase-3 inhibition promotes viral latency.
Materials and Methods
Chemicals and Reagents
Rabbit polyclonal/mouse monoclonal antibodies against Alix, AKT, APOBEC3G, Brd4, CD3, CDK9, gp130, IRAK4, IRF3, IRF7, Jak2, MyD88, PI3K, PLCγ1, SP1, Trim5a, TLR1, Tsg101 and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal antibodies againstAP-1, Cyclin T1, Erk1/2, NFAT, NF-kB p65, p38, PCAF, p-Rpb-CTD, STAT1, STAT3, STAT5, TCRβ, YB-1, vif and ZAP70 were bought from Cell Signaling Technology, Inc (Danvers, MA). Caspase-3 fmk Inhibitor Z-DEVD was obtained from R and D systems (Minneapolis, MN). caspase-3 activator PAC1 and all other chemicals were from Sigma (St. Louis, MO).
Cell Culture and Treatments
A3.01 (CEM A3.01 (RRID:CVCL-6244)) cells were obtained from the National Institutes of Health AIDS Research Reference and Reagent Program (Germantown, MD) and cultured at 37°C in 5% CO2 in RPMI 1640 medium containing 10% fetal calf serum, 2 mM glutamine, 50μg/ml penicillin, and 50μg/ml streptomycin.
To perform HIV-1 infection, cells were seeded at 2 X 105 cells/ ml for 24 h, infected with known amounts of HIV-1 (MN, 109 copies per 106 cells) for 2 h, washed twice with PBS, and cultured for 3 days. Cells were then incubated with 50 μM of Caspase-3 fmk Inhibitor Z-DEVD, or 25 nMof caspase-3 activator PAC1 for another 7 days and cultured for 7 additional days after fresh medium was added (Supp. Figure A).
RNA Isolation and DNA Isolation
Viral RNA was isolated from 140 μl of culture supernatant using the QIAamp Viral RNA Mini Kit (Valencia, CA 91355) according to the manufacturer’s protocol. 5 out of the 50 μl of the extracted RNA were used as templates for real-time RT-PCR. Known concentrations of HIV-1 (MN) viral RNA (serially diluted: 108 to 100 copies) were used as templates. Quantitative RT-PCR was performed to generate a standard curve. Each value shown in the figures represents the average concentration of 6 reactions in triple isolated repeats based on the standard curve.
Total DNA isolation of approximately 1 x 107 cells was performed with the QIAamp DNA Mini Kit (Valencia, CA 91355) according to the manufacturer’s protocol. Five out of 100 μl of the extracted DNA was used as templates for real-time PCR. Known concentrations of HIV-1 (MN) viral DNA (serially diluted: 108 to 100 copies) were used as templates. Quantitative PCR was performed to generate a standard curve. Each value shown in the figures represents the average concentration of 6 reactions in triple isolated repeats based on the standard curve.
Real-time PCR
Primers and TaqMan probes were designed for the gag p24, which is the variable region among most of the HIV-1 subtype B isolate sequences according to the Gen Bank database. The forward primer was 5’-GACATCAAGCAGCCATGCAA-3’, corresponding to nucleotides 1367–1386, and the reverse primer was 5’-CTATCCCATTCTGCAGCTTCCT-3’, corresponding to nucleotides 1430–1409. The Taq-Man probes were composed of the oligonucleotide sequence 5’-ATTGATGGTCTCTTTTAACA-3’, corresponding to nucleotides 1488–1507, coupled with a reporter dye [6-carboxy fluorescein] (FAM) at the 5’ end and a non-fluorescent quencher and a Minor Groove Binder (MGB), which is a Tm enhancer, at the 3’ end. The nucleic acids were amplified and detected in an automated TaqMan 7500 Analyzer using QuantiTectTM Probe RT-PCR kit (Qiagen Inc., Valencia, CA). The 25 μl PCR mixture consisted of 100 nM primers and 100 nM probe and underwent the following conditions: 95° C for 10 min, 45 cycles of two-step PCR at 95° C for 15 s and at 60° C for 1 min.
For RT-PCR, the following thermal steps were performed: 55° C for 5 min, at 50° C for 30 min and at 95° C for 10 min, 45 cycles of two-step PCR at 95° C for 15s and at 60° C for 1 min.
Western Blot Analysis
Proteins were isolated from A3.01 cells with RIPA buffer (1×PBS, 1% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 0.1 mg/ml PMSF, 30 μl/ml aprotinin, 1 mM sodium orthovanadate). Equal amounts of protein were boiled in the loading buffer (100 mM Tris–HCl, 200 mM DTT, 4% SDS, 0.2% bromphenol blue, 20% glycerol), separated on SDS-PAGE and blotted onto polyvinylidene difluoride membranes. Data represented are from three independent experiments. The relative quantitation of protein expression was determined using Image J (Image Processing and Analysis in Java) from NIH website (https://imagej.nih.gov/ij/).
Statistical Analysis
The unpaired Student’s t test was used for data analyses as indicated, and p-value < 0.05 (*) and p-value < 0.01 (**) were considered significant and very significant, respectively.
Results
After infection with HIV-1 for 3 days, the A3.01 cells were incubated in medium containing 50 μm of Caspase-3 fmk Inhibitor Z-DEVD, or 25 nm of caspase-3 activator PAC1 for an additional14 days (Supp. Figure A). Cells were tested for caspase-3 activities with EnzChek® Caspase-3 Assay Kit #1, Z-DEVD-AMC Substrate. We found that on day 14, caspase-3 activities were inhibited by caspase inhibitor, while the activator increased caspase-3 activities significantly (Supp. Figure B).
The Effects of Caspase-3 on HIV-1 Latency
Previously, we reported that when Jurkat cells were treated with gp120 or infected with HIV-1 and HIV-2, both Fas/FasL- and Bax/ mitochondria-mediated apoptotic signaling pathways were induced with increased caspase-1 activities [3,4]. To investigate whether caspase-3 could affect HIV-1 infection and replication, we infected A 3.01 cells with HIV-1 for 3 days, followed by incubation in medium containing caspase-3 inhibitor or activator for another 14 days. Caspase-3 activator treatment led to significant increases in viral RNA yields in the supernatants of cell cultures, relative to the DMSO control (Figs. 1A, shaded dark bars). In contrast, caspase-3 activator significantly decreased the amount of viral DNA relative to DMSO control (Figs. 1A, unshaded white bars), suggesting that caspase-3 activation could result in decreased reverse transcription activity and viral DNA integration, potentially in favor of viral replication and propagation.Caspase-3 inhibitor decreased HIV-1 replication as expected, consistent with our previous work [8,15], which confirms that the amount of HIV-1 replication is inversely proportional to its viral DNA level (Fig. 1A, unshaded white bars).Taken together, these data demonstrate that inhibition of apoptosis potentially attenuates HIV-1 latent reservoir seeding (in favor of active replication) while activation of apoptosis, in contrast, may potentiate this mechanism.
It is well known that the HIV-1 LTR (long terminal repeat) has recognition sequences for many cellular transcription factors, including NFAT (Nuclear Factor of Activated T-Cells), NF-κB (nuclear factor κB), AP-1 (activator protein 1) and SP-1 (specificity protein 1) which play important roles in HIV-1 replication [18,19]. In order to examine whether caspase-3 affects the expression of these host transcription factors, total proteins from cell pellets were analyzed by Western blot to detect NF-κB p65, Ap-1, Sp-1 and NFAT. As shown in (Figure 1B), caspase-3 inhibitor dramatically down regulated expression of NF-κB p65, Ap-1, Sp-1 and NFAT (Figure 1B). In contrast, the caspase-3 activator significantly upregulated expression of NF-κB p65, Ap-1, Sp-1 and NFAT (Figure 1B).
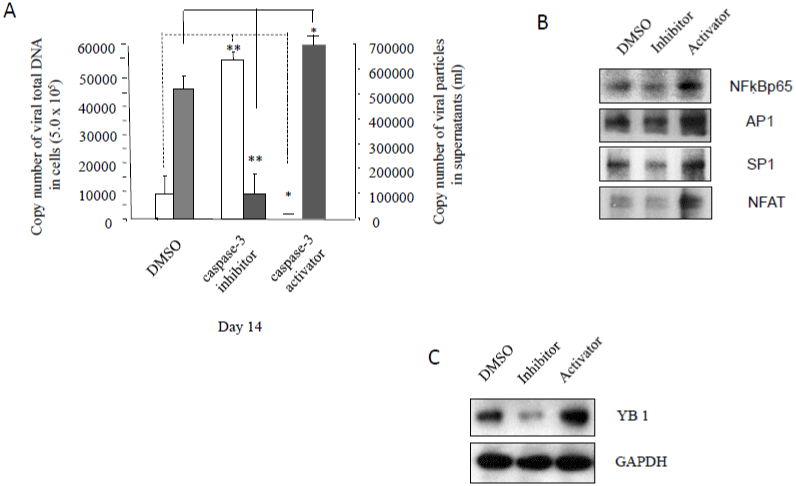
Figure 1: The effects of caspase-3 activities on HIV-1 latency: A3.01 cells, infected with HIV-1 (MN) for 3 days, were incubated in medium containing caspase-3
inhibitor, Z-DEVD, or caspase-3 activator, PAC1, for 14 days, and DMSO was used as control. (A).140 μl of culture supernatants containing HIV-1 particles and
were used to isolate viral RNA for RT-PCR to test viral replication. 1 x 107 of live cells were used to isolate viral DNA for PCR to detect viral DNA levels. Total cell
lysates were subjected to Western blot analysis to detect (B) NF-κB p65, Ap-1, Sp-1 and NFAT; and (C) YB-1.
The DNA and RNA binding protein Y-box-binding Protein 1 (YB-1), also known as Y-box transcription factor, plays an important role as a cofactor to support early and late steps of HIV replication and YB-1 depletion resulted in a 10-fold decrease in HIV-1 replication in different cell lines [20]. We found that caspase-3 inhibitor dramatically deceased YB-1 expression, while caspase-3 activator increased YB-1 expression (Figure 1C).
These data indicate that inhibition of caspase-3 can decrease HIV- 1 replication and increase virial latency by down regulated expression of host transcription factors.
Caspase-3 Inhibitor Suppressed T-Cell Receptor (TCR)- Related Signaling Pathways and MAPK Pathways
Latent HIV proviruses are thought to be primarily reactivated in vivo through stimulation of the T-cell receptor (TCR). It has been reported that T cell activation is required forHIV-1 replication and gene expression, and T cells are generally activated through TCR, and CD3 [19,21], or/and PI3K/Akt [19]-related cell signaling pathways. These pathways can trigger and activate downstream molecules, such as Zap-70 and PLCγ1, and serially activate transcription factors, NFκB p65, Ap-1, Sp-1 and NFAT. Total cell lysates were analyzed by Western blot. As shown in Fig. 2, caspase-3 inhibitor could down regulate the expression of receptors CD3, and TCRβ (Figure 2A), and decrease protein expression of Zap-70, and PLCγ1 (Figure 2A). Caspase-3 inhibitor also reduced PI3K/Akt expression (Figure 2B) in A3.01 cells. In contrast, caspase-3 activator was able to increase the expression of CD3, TCRβ, Zap-70 and PLCγ1 (Figure 2A), and PI3K/ Akt (Figure 2B). These results indicate that the inhibition of caspase-3 can inactivate T cells through TCR-related signaling pathways, which are required for T cell activation and viral replication.
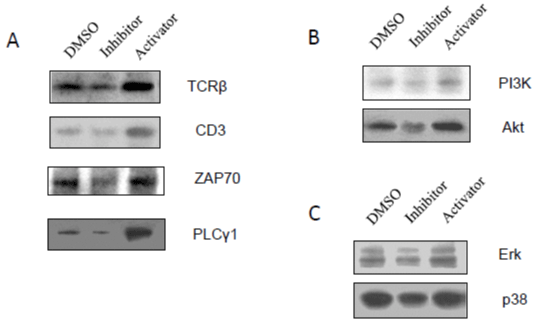
Figure 2: The effects of caspase-3 activities on T-cell receptor (TCR)-related signaling pathways and MAPK pathways: A3.01 cells, infected with HIV-1
(MN) for 3 days, were incubated in medium containing caspase-3 inhibitor, Z-DEVD, or caspase-3 activator, PAC1, for 14 days, and DMSO was used as control.
Total cell lysates were subjected to Western blot analysis to detect (A) TCRβ, CD3, ZAP 70 and PLCγ1; (B) PI3K and Akt; and (C) Erk and p38.
MAPK (mitogen-activated protein kinase) is another type of signaling pathway that plays an important role in activation of host transcription factors, such Ap-1, and NF-κB [18,19,22]. To elucidate the MAPKs involved in the effects of caspase-3 on HIV-1 latency, total cell lysates were analyzed by Western blot to detect MAPK, Erk1/2 and p38. As shown in (Figure 2C), caspase-3 inhibitor was able to decrease the expression of Erk1/2 and p38 relative to DMSO control. These results suggest that caspase-3 inhibition increases HIV-1 latency at the viral transcriptional levels with blockage of host transcription factors, which are required for viral RNA production by inactivating TCR-related/MAPK signaling pathways.
Caspase-3 Inhibitor Blockedp-Tefb Signaling Pathways
The recruitment of P-TEF b (positive Transcription Elongation Factor b) by Brd4 (Bromodomain containing protein 4) during the elongation stage is very important for full-length synthesis of HIV- 1 mRNA, which is regulated by the Tat protein essential for viral replication. The protein cyclin T1 is tightly associated with CDK9 (Cyclin-Dependent Kinase 9) to form a major subunit of P-TEFb [23]. The CDK9/cyclin T1 complex is a key factor for productive elongation of transcription of HIV genoms. It was found that caspase-3 inhibitor could dramatically decrease the protein expression of Brd4, CDK9, and cyclin T1 (Figure 3A).
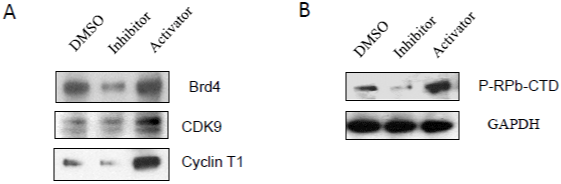
Figure 3: The effects of caspase-3 activities on p-TEFb signaling pathways: A3.01 cells, infected with HIV-1 (MN) for 3 days, were incubated in medium
containing caspase-3 inhibitor, Z-DEVD, or caspase-3 activator, PAC1, for 14 days, and DMSO was used as control. Total cell lysates were subjected to Western
blot analysis to detect (A) Brd4, CDK9 and Cyclin T1; and (B) P-RPb-CTD.
Transcription of many viruses is dependent on host cell factors such as RNA polymerase II (pol II) which is made up of 12 subunits. The Carboxyl-Terminal Domain (CTD) of the largest subunit of pol II plays a central role in transcriptional and co-transcriptional RNA processing. CTD modification generates a code that regulates interaction with transcription and RNA processing factors [23,24]. Among the CTD modifications, the phosphorylation of Ser2 (Ser2 P) of pol II CTD, is catalyzed by CDK 9 subunit of P-TEFb [14,23,24]. We found that A3.01 cells treated withcaspase-3 inhibitor displayed decreased phosphorylated pol II CTD (Figure 3B).
In contrast, caspase-3 activator was able to induce P-TEFb signaling pathways through upregulation of proteins, Brd4, CDK9 and Cyclin T1 (Figure 3A), which induce the elongation of HIV-1 transcription, and activation of pol II CTD (Figure 3B) required for HIV-1 transcription and elongation in A 3.01 cells.
Caspase-3 Inhibitor Inactivatedgp130/Jak-Related Signaling Pathways
Glycoprotein 130 (gp130), a transmembrane protein with no intrinsic tyrosine kinase activity, can be phosphorylated on tyrosine residues by other proteins. Phosphorylation leads to association with JAK and STAT protein transcription factors and/or activates other pathways including STAT3-MAPK signaling [25]. It was reported that activation of Jak-STAT pathways correlated with upregulating the levels of integrated viral HIV DNA and HIV replication [26]. The Jak-STAT pathways induced/activated by p38a could resist action of AZT in inhibition of HIV-1 replication [27]. To investigate whether the gp130-Jak-STAT signaling pathways are involved in the effects of caspase-3 on HIV-1 latency, total cell lysates were analyzed by Western blotting to detect gp130, Jak, and STATs. As shown in (Figure 4), caspase-3 inhibitor could reduce the expression of gp130 and Jak2 (Figure 4A), Stat5, Stat3 and Stat1 (Figure 4B). In contrast, caspase-3 activator increased expression of gp130 and Jak2 (Figure 4A), Stat5, Stat3 and Stat1 (Figure 4B) to increase viral RNA production.
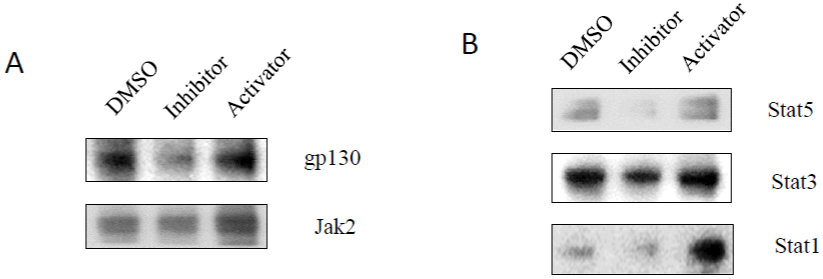
Figure 4: The effects of caspase-3 activities on gp130/Jak-related signaling pathways: A3.01 cells, infected with HIV-1 (MN) for 3 days, were incubated in
medium containing caspase-3 inhibitor, Z-DEVD, or caspase-3 activator, PAC1, for 14 days, and DMSO was used as control. Total cell lysates were subjected to
Western blot analysis to detect (A) gp130 and Jak2; and (B) STAT5, STAT3 and STAT1.
Caspase-3 Inhibitor Inhibited MyD88-Related Signaling Pathways
The MyD88 protein (myeloid differentiation primary response 88) acts as an adaptor, connecting proteins that receive signals from outside the cell to the proteins that relay signals inside the cell. In innate immunity, the MyD88 plays a pivotal role in immune cell activation through Toll-like receptors (TLRs), which are integral membrane glycoproteins with typical semicircular-shaped extracellular parts containing leucine-rich repeats responsible for lig and binding, and intracellular parts containing Toll-Interleukin receptor (TIR) domain [28]. MyD recruits IL-1 receptor-associated kinase-4 (IRAK-4) to TLRs through interaction of the death domains of both molecules [28]. MyD88-related signaling pathways then activate NF-κB, and IRFs (interferon-regulatory factor) [29]. IRFsis a group of proteins responsible for expression of type I interferons setting up the antiviral state of a cell [28,29]. The activation of MyD88-mediated pathways was reported to induce HIV-1 replication and reactivate viral production from latency [30]. We found that caspase-3 inhibitor could down regulate expression of MyD88 (Figure 5A), TLR1 (Figure 5B), IRAK-4 (Figure 5C), and IRF-7 and IRF-3 (Figure 5D). These data indicate that inhibition of caspase-3 can decrease HIV- 1 replication and increase viral DNA production by blockage of My D88-meidated signaling pathways.
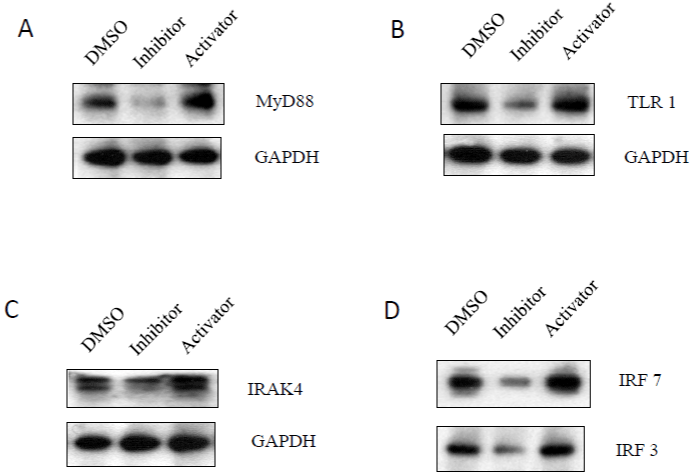
Figure 5: The effects of caspase-3 activities on MyD88-related signaling pathways: A3.01 cells, infected with HIV-1 (MN) for 3 days, were incubated in
medium containing caspase-3 inhibitor, Z-DEVD, or caspase-3 activator, PAC1, for 14 days, and DMSO was used as control. Total cell lysates were subjected to
Western blot analysis to detect (A) MyD88; (B) TLR1; (C) IRAK4; and (D) IRF7 and IRF7.
Caspase-3 Activities Affected Epigenetic Regulator, PCAF
Cellular epigenetic factors can regulate HIV latency and reactivation by affecting the chromatin state in the vicinity of the viral promoter located to the 5’ Long Terminal Repeat (LTR) sequence [31]. Some modifications via methylation and acetylation at a particular residue of histone tails can alter accessibility of transcription factors and viral RNA polymerizing machinery to the HIV-1 5' LTR [14]. Chromatin acetylationby histone acetyl transferases (HATs) promotes unfolding of chromatin, is associated with active euchromatin and can increase viral production. P300/CBP-Associated Factor (PCAF) is a human gene and transcriptional co-activator associated with p53. p300 (histone acetyltransferase p300) serves as histone acetyl transferases to transfer acetylation at residues of histone 3 and PCAF is involved in reactivating HIV-1 replication from latency [32]. As shown in (Figure 6), caspase-3 inhibitor deceased PCAF expression, while caspase-3 activator increased PCAF expression.
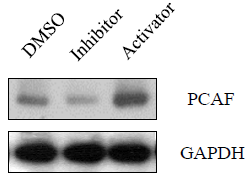
Figure 6: The effects of caspase-3 activities on the expression of
epigenetic regulator, PCAF. A3.01 cells, infected with HIV-1 (MN) for 3
days, were incubated in medium containing caspase-3 inhibitor, Z-DEVD, or
caspase-3 activator, PAC1, for 14 days, and DMSO was used as control.
Total cell lysates were subjected to Western blot analysis to detect PCAF.
Caspase-3 Activities Modulated Viral Restriction Factors, TRIM5a and APOBEC3G, and HIV-1 Budding
TRIM5a is a retrovirus restriction factor which mediates a species-specific, early block to retrovirus infection.TRIM5a, which binds HIV-1 p24 capsid protein, mediates uncoating of incoming viral cores and blocks retroviral replication [33]. Caspase-3 inhibitor could increase expression of TRIM5a, while caspase-3 activator could decrease TRIM5a expression (Figure 7A), suggesting that inhibition of caspase-3 activities may block viral RNA uncoating and reverse transcription.
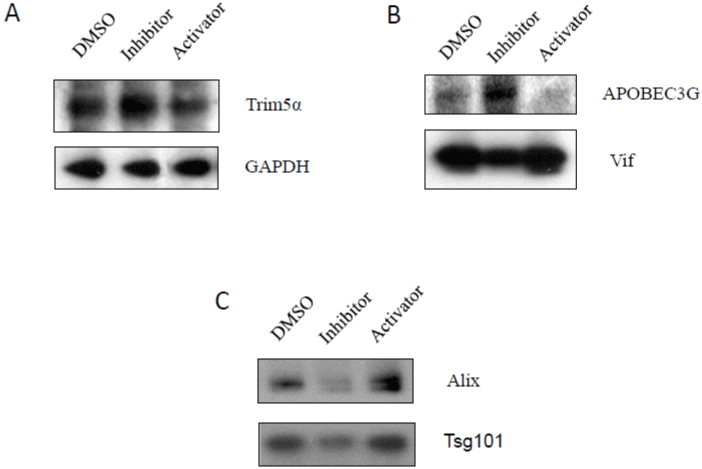
Figure 7: The effects of caspase-3 activities on the expression of viral restriction factors, TRIM5a and APOBEC3G, and HIV-1 budding: A3.01 cells,
infected with HIV-1 (MN) for 3 days, were incubated in medium containing caspase-3 inhibitor, Z-DEVD, or caspase-3 activator, PAC1, for 14 days, and DMSO
was used as control. Total cell lysates were subjected to (A) Trim5a; (B) APOBEC3G; and (C) Tsg101 and Alix.
APOBEC3G is a member of a large family of cytosine deaminases that is able to convert cytosine to uracil during reverse transcription in the next round of HIV-1 infection, which leads to G-to-A hypermutation in the newly synthesized viral DNA, potentially leading to its instability [33,34]. APOBEC3G also induces defects in reverse transcription and DNA integration [34]. Caspase-3 inhibitor could increase expression of APOBEC3G, while caspase-3 activator could decrease APOBEC3G expression (Figure 7B). Viral infectivity factor (vif) is a protein expressed by HIV-1, which is essential for viral replication [34]. Vif inhibits the activities of APOBEC3G [27]. Caspase-3 inhibitor could decrease expression of vif, while caspase-3 activator could increase vif expression (Figure 7B), suggesting that caspase-3 inhibition can increase the risk of viral mutation caused by APOBEC3G, and leading to viral instability and decreased viral production.
It is known that retroviral budding depends on the engagement of a complex network of host endosomal sorting proteins that include TSG101 and Alix in ESCRT (endosomal sorting complex required for transport) pathway [35, 36]. Tsg101 plays an important role in the pathogenesis of HIV [35,36], and HIV-1 Gag is able to bind and recruit the Tsg101 protein to gain access to the downstream machinery and to mediate viral budding [10,35,36]. Alix encodes a multimerization activity that is essential for Alix-dependent HIV-1 release [35,36]. Caspase-3 inhibitor could decrease expression of Alix and Tsg101, while caspase-3 activator could increase expression of Alix and Tsg101 (Figure 7C). These results indicate that inhibition of caspase-3 down regulates expression of Tsg101 and Alix, which may result in inhibition of HIV-1 budding and viral maturation.
Discussion
Recently, we reported that proapoptotic components from apoptotic signaling pathways promote HIV-1 replication (8) and inhibit HIV-1 latency (15) while antiapoptotic molecules decrease HIV-1 replication (8) and increase HIV-1 latency [15]. Caspase-3 is a caspase protein that interacts with caspade-8 and caspase-9. The catalytic site of caspase-3 involves the sulfohydryl group of Cys- 163 and the imidazole ring of His-121 [37]. In vitro, caspase-3 has been found to prefer the peptide sequence DEVDG (Asp-Glu- Val-Asp-Gly) with cleavage occurring on the carboxy side of the second aspartic acid residue (between D and G) [37]. Caspase-3 is activated in the apoptotic cell both by extrinsic (death lig and) and intrinsic (mitochondrial) pathways.It has been reported that HIV-1 replication can be induced by super infection with HIV-1 strains [22] or co-infection with other viruses [38] through activation of apoptotic pathways and increased caspase-3 activities. Inactivating caspase-3 activities inhibits HIV-1 replication and promotes HIV-1 latency [15] (Figure 1A), while activating caspase-3 activities promotes HIV- 1 replication and inhibits HIV-1 latency (Figure 1A).
Evidence from literature shows that virus latency does not require a complete shutdown of viral gene expression, but only the absence of infectious progeny production [14,19]. Due to latent reservoir, ART is not curative, and temporarily suppresses HIV gene expression and replication. If the therapy is interrupted or drug resistance occurs, it will be result in a rapid viral rebound arising from latency and re-enter to acute infection stage [14,19]. Several mechanisms can silence HIV replication and the production of infectious viral particles by ART treatment, such as deleterious mutations in the viral genome, transcriptional interference, changes in chromatin structure/epigenetic silencing, and problems with RNA processing and transport [13,14].
I It has been reported that anti-apoptotic molecules, such as FLIP [10] and Bcl2 [39], or knock-down expression of proapoptotic component, FADD [9], inhibits HIV-1 replication and promotes latency in many ways/levels.
Upon HIV-1 infection, viral particles bind to CD4, the HIV receptor, in the raft region on the surface of host cells, and the viral core, in which the viral RNA genome is enclosed by a capsid shell, enters the cells. In order to reverse transcribe into viral cDNA, theviral RNA is required to be released from the capsid shell, called uncoating [10]. TRIM5a is a cellular protein that binds to determinants present on a retroviral capsid soon after the entry of the viral core into the cytoplasm of a target cell and resistant against uncoating of viral RNA [33]. FLIP can inhibit the binding of viral particles to CD3 [10], andboth FLIP [10] and inhibition of caspase-3 activities (Figure 7A) can inhibit HIV-1 uncoating with increased expression of TRIM5a in lymphocytes.
APOBEC3G is a member of the cellular polynucleotide cytidine deaminases, which catalyze the deamination of cytosine (dC) to uracil (dU) in single-stranded DNA. These enzymes potently inhibit the replication of a variety of retroviruses and retrotransposons, including HIV-1 [33,34]. It is possible those both FLIP [10] and inhibition of caspase-3 activities (Figure 7B) play roles in increased deleterious mutations in the viral genome with increased expression of APOBEC3G and decreased HIV-1 vif expression.
Host transcription factors, such NF-κB p65, Ap-1, Sp-1 and NFAT, with binding sites located within the viral Long Terminal Repeat (LTR), are required for HIV-1 gene expression and replication which can be regulated by activation/inhibition of TCR-related signaling pathways [9,10,17,19,21]. Knock-down of the expression of FADD [9] and inhibition of caspase-3 activities (Figure 1B) inactivates NF-κB p65, Ap-1, Sp-1 and NFAT through decreased/ inactivated signaling transduction of TCR-related pathways with down regulation of expression of TCRa/β, PKC, or PI3K/Akt [9] (Figure 2). In addition to transcriptional interference with NF-κB p65, Ap-1, Sp-1 and NFAT by anti-apoptotic activities [9,10] (Figure 1), other transcription factors, such nuclear factor 1 (NF-1), IRFs, and STATs, are also involved in HIV-1 transcription, in which FLIP expression decreases NF-1C expression [10]. Inhibition of caspase-3 activities down regulates IRF3 and IRF7 through MyD88-related signaling pathways (Figure 5) and STAT1/3/5 through gp130/Jak signaling pathways (Figure 4).
HIV-1 transcriptional transactivator (Tat) is essential for synthesis of full-length transcripts from the integrated viral genome by RNA polymerase II (Pol II). Tat recruits the host positive transcription elongation factor b (P-TEFb) to the HIV-1 promoter through binding to the transactivator RNA (TAR) at the 5’-end of the nascent HIV transcript [23,24]. P-TEFb functions to promote the transition of RNA polymerase II from abortive to productive elongation [24]. Inhibition of caspase-3 activities may hinder the full length of synthesis of HIV-1 mRNA by inhibition of P-TEFb pathways (Figure 3) at the HIV-1 mRNA transcription level.
Accumulated evidence verifies that epigenetic regulation, i.e., histone modifications via methylation and acetylation, plays an important role in the establishment and maintenance of HIV- 1 latency [14,31]. Generally, chromatin acetylation by Histone Acetyl Transferases (HATs) promotes chromatin opening and the accessibility of the nucleosomal DNA to transcription factors [14,31,32]. The inhibition of caspase-3 activities may control the changes in chromatin structure through decreased expression of PCAF (Figure 6), which can serve as HAT to loosen the structure of nucleosome to allow RNA polymerase II to contact and bind to viral DNA easily.
The ESCRT (endosomal sorting complex required for transport) pathway is a key mediator of MVB (Multivesicular body) biogenesis, but it also plays critical roles in retroviral budding, including HIV- 1 [35,36]. Many studies have indicated that HIV-1 usurps ESCRT machinery [35,36] to exit the cell. FLIP can block the trafficking of gp120 and Gag p24 capsid protein into lipid rafts with inhibition of the Tsg101 and Alix in ESCRT signaling pathway [10]. Knock-down of FADD [9] and inhibition of caspase-3 activities (Figure 7C) can decrease the expression of Tsg101 and Alix, and knock-down of FADD can also block the translocation of Tsg101 and Alix into lipid raft [9], where the viral budding occurs.
Once inside the cytoplasm, reverse transcriptase synthesizes double stranded DNA from the viral RNA genome. For integration to occur in the nucleus, the virus must successfully cross the nuclear membrane [40]. HIV-1 establishes the Pre-integration Complex (PIC) that is responsible for nuclear import [40]. After PIC enters the nucleus, the viral DNA can undergo two different pathways [41]. In one pathway, the viral genome circularizes by integrating onto itself. Recently it has reported that unintegrated HIV-1 DNA can be rescued to transcript to RNA and express viral genes [42]; though there exist controversies if the unintegrated viral DNA by NF-κB can produce any infective virions [41,42]. The second pathway is to integrate its genome into the host, which leads to latent infection. In addition, clinically, total HIV DNA load in blood, tissues, and cells provides insights into HIV pathogenesis, probably because all viral forms participate in host cell activation and HIV pathogenesis [43]. Total HIV DNA is thus a biomarker of HIV reservoirs [43]. Therefore, in this study the total genomic DNA, including unintegrated viral DNA and integrated viral DNA, was used to determine to compare the size of latent reservoirs with each other. We have found that the larger HIV-1 reservoirs (more viral total DNA production), the more HIV- 1 replication during reactivation [15].
Several clinical studies reported that the proviral HIV-1 DNA copy number on ART is directly related to the level of persistent immune activation [44], although contradictory results show that there is no relationship of HIV-1 DNA copy number with immune activation [44,45]. In our experiment condition, HIV-1 latency is inversely proportional to viral replication.Further studies are needed to elucidate the mechanism of how components of the apoptotic pathways modulate the seeding of latent reservoirs at DNA level, and how the antiapoptotic activities affect the pre-integration complex (PIC) with inserting a larger amount of viral DNA to host chromosome DNA relative to the effect of proapoptotic molecules.
In conclusion, differential modulation of caspase-3 activities can affect the status of HIV-1 latency, leading to decreased/increased viral latency marketed with differential products of viral DNA and viral RNA.Caspase-3 inhibitor promotes viral latency in Jurkat cells [15] and A3.01 cells at the different stages of HIV-1 infection and replication. Caspase-3 inhibition may inhibit viral RNA uncoating normally by increasing Trim5a expression and may increase the chance of viral cDNA mutation (G to A) with upregulation of APOBEC3G. Caspase-3 inhibition may decrease viral RNA transcription from genome of host cells through down regulation of NF-κB p65, Ap-1, Sp-1 and NFAT in TCR/PI3K-Akt-related pathways; STAT1, STAT3 and STAT5 in gp130-Jak pathways; IRF3 and IRF7 in MyD88-related pathways; and inactivation of YB-1 and MAPK, ERK and p38, activity. Caspase-3 inhibition may hinder the full length of HIV-1 mRNA synthesis through inhibition of p-TEFb signaling pathways with down regulation of Brd4, CDK9, Cyclin T1 and p-RPb-CTD. Caspase-3 inhibition down regulates PCAF, which serves as Histone Acetyl Transferases (HATs) for chromatin acetylation, leading to blockage of reactivating HIV-1 replication from latency. The diminishing of the protein level of Tsg101 and Alix by caspase-3 inhibition may block trafficking of the components of viral particle through ESCRT signaling system from cytoplasm to plasma membrane where viral budding occurs. Our study clearly demonstrates the complex interplay of host factors and apoptotic molecules in the regulation of cell death by HIV-1 infection and latency which may provide new insights and targets for cure of HIV infection.
Author Contributions
XW and IH conceived the project. XW, HH, SB, JZ, and KD performed the experiments. XW formatted datasets and analyzed data. XW and IH participated in manuscript writing, data interpretation and contributed to the intellectual content of this work.
Acknowledgments
The authors wish to acknowledge of Dr. Viswanath Ragupathy and Dr. Yiden Liang for their critical review of this manuscript. The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
Competing Interests
The authors declare no competing interests.
References
- Global HIV & AIDS statistics — Fact sheet.
- Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, et al. Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive antiretroviral therapy. J Neuroimmune Pharmacol. 2019; 14: 110-9.
- Wang X, Viswanath R, Zhao J, Tang S, Hewlett I. Changes in the level of apoptosis-relatedproteins in Jurkat cells infected with HIV-1 versus HIV-2. Mol Cell Biochem. 2010; 337: 175–183.
- Wang X, Zhao J, Tang S, Lee S, Glazer RI, Hewlett I. I.C-FLIPL regulates PKC via AP-2 to inhibit Bax-mediated apoptosis induced by HIV-1 gp120 in Jurkat cells. Mol Cell Biochem. 2009; 330: 23-9.
- Timilsina U, Gaur R. Modulation of apoptosis and viral latency - an axis to be well understood for successful cure of human immunodeficiency virus. J Gen Virol. 2016; 97: 813-24.
- Garg H, Joshi A. Host and Viral Factors in HIV-Mediated Bystander Apoptosis. Viruses. 2017; 9: 237-59.
- Majumder B, Venkatachari NJ, Srinivasan A, Ayyavoo V. HIV-1 mediated immune pathogenesis: spotlight on the role of viral protein R (Vpr). Curr HIV Res. 2009; 7: 169-77.
- Wang X, Ragupathy V, Zhao J, Hewlett I. Molecules from apoptotic pathways modulate HIV-1 replication in Jurkat cells. Biochem Biophys Res Commun. 2011; 414: 20-4.
- Wang X, Tan J, Zhao J, Ragupathy V, Haleyurgirisetty M, Hewlett I. Some findings of FADD knockdown in inhibition of HIV-1 replication in Jurkat cells and PBMCs. Mol Cell Biochem. 2014; 393: 181-90.
- Tan J, Wang X, Devadas K, Zhao J, Zhang P, Hewlett I. Some mechanisms of FLIP expression in inhibition of HIV-1 replication in Jurkat cells, CD4+ T cells and PBMCs. J Cell Physiol. 2013; 228: 2305-13.
- Bren GD, Whitman J, Cummins N, Shepard B, Rizza SA, Trushin SA, et al. Infected cell killing by HIV-1 protease promotes NF-kappaB dependent HIV-1 replication. PLOS ONE. 2008; 3: e2112.
- Bren GD, Trushin SA, Whitman J, Shepard B, Badley AD. HIV gp120 induces, NF-kappaB dependent, HIV replication that requires procaspase 8. PLOS ONE. 2009; 4: e4875.
- Khoury G, Darcis G, Lee MY, Bouchat S, Van Driessche B, Purcell DFJ, et al. The molecular biology of HIV latency. Adv Exp Med Biol. 2018; 1075: 187-212.
- Khanal S, Schank M, El Gazzar M, Moorman JP, Yao ZQ. HIV-1 latency and viral reservoirs: existing reversal approaches and potential technologies, targets, and pathways involved in HIV latency studies. Cells. 2021; 10: 475- 97.
- Wang X, Zhao J, Biswas S, Devadas K, Hewlett I. Components of apoptotic pathways modulate HIV-1 latency in Jurkat cells. Microbes Infect. 2021; 24: 104912.
- Khan SZ, Hand N, Zeichner SL. Apoptosis-induced activation of HIV-1 in latently infected cell lines. Retrovirology. 2015; 12: 42.
- Wang X, Zhao J, Mbondji C, Hewlett I. p53 expression activation of HIV-1 latency in U1 cells. Int J Virol AIDS. 2017; 3: 036.
- Al-Harthi L, Roebuck KA. Human immunodeficiency virus type-1 transcription: role of the 5'-untranslated leader region [review]. Int J Mol Med. 1998; 1: 875- 81.
- Pluta A, Jaworski JP, Cortés-Rubio CN. Balance between retroviral latencyand transcription: based on HIV Model. Pathogens. 2020; 10: 16-41.
- Weydert C, van Heertum B, Dirix L, De Houwer S, De Wit F, Mast J, et al. Y-box-binding protein 1 supports the early and late steps of HIV replication. PLOS ONE. 2018; 13: e0200080.
- Carlin E, Greer B, Lowman K, Duverger A, Wagner F, Moylan D, et al. Extensive proteomic and transcriptomic changes quench the TCR/CD3 activation signal of latently HIV-1 infected T cells. PLOS Pathog. 2021; 17: e1008748.
- Wang X, Sun B, Mbondji C, Biswas S, Zhao J, Hewlett I. Differences in activation of HIV-1 replication by superinfection with HIV-1 and HIV-2 in U1 cells. J Cell Physiol. 2017; 232: 1746-53.
- Fujinaga KP-TE. P-TEFb as A promising therapeutic target. Molecules. 2020; 25: 838-365.
- Jiang L, Huang Y, Deng M, Liu T, Lai W, Ye X. Polo-like kinase 1 inhibits the activity of positive transcription elongation factor of RNA Pol II b (P-TEFb). PLOS ONE. 2013; 8: e72289.
- Schumertl T, Lokau J, Rose-John S, Garbers C. Function and proteolytic generation of the soluble interleukin-6 receptor in health and disease. Biochim Biophys Acta Mol Cell Res. 2022; 1869: 119143.
- Gavegnano C, Brehm JH, Dupuy FP, Talla A, Ribeiro SP, Kulpa DA, et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLOS Pathog. 2017; 13: e1006740.
- Wang X, Zhao J, Ragupathy V, Hewlett I. The effects of MAPK p38alpha on AZT resistance against reactivating HIV-1 replication in ACH2 cells. Mol Cell Biochem. 2019; 462: 41-50.
- Ahmed H, Khan MA, Kahlert UD, et al. Role of adaptor protein myeloid. Differentiation 88 (MyD88) in Post-Subarachnoid Hemorrhage Inflammation: A Systematic Review. Int J Mol Sci. 2021; 22: 4185-98.
- Cui J, Chen Y, Wang HY, Wang RF. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccin Immunother. 2014; 10: 3270-85.
- Ariza ME, Ramakrishnan R, Singh NP, Chauhan A, Nagarkatti PS, Nagarkatti M. Bryostatin-1, a naturally occurring antineoplastic agent, acts as a toll-like receptor 4 (TLR-4) ligand and induces unique cytokines and chemokines in dendritic cells. J Biol Chem. 2011; 286: 24-34.
- Koutelou E, Farria AT, Dent SYR. Complex functions of Gcn5 and Pcaf in development and disease. Biochim Biophys Acta Gene Regul Mech. 2021; 1864: 194609.
- Pan C, Mezei M, Mujtaba S, Muller M, Zeng L, Li J, et al. Structure-guided optimization of small molecules inhibiting human immunodeficiency virus 1 Tat association with the human coactivator p300/CREB binding proteinassociated factor. J Med Chem. 2007; 50: 2285-8.
- D’Urbano V, Bertoldi A, Re MC, De Crignis E, Tamburello M, Primavera A, et al. Restriction Factors expression decreases in HIV-1 patients after cART. New Microbiol. 2021; 44: 95-103.
- Stupfler B, Verriez C, Gallois-Montbrun S, Marquet R, Paillart JC. Degradation-independent inhibition of APOBEC3G by the HIV-1 Vif protein. Viruses. 2021; 13: 617-33.
- Balasubramaniam M, Freed EO. New insights into HIV assembly and trafficking. Physiology (Bethesda). 2011; 26: 236-51.
- Mouhand A, Pasi M, Catala M, Zargarian L, Belfetmi A, Barraud P, et al. Overview of the nucleic-acid binding properties of the HIV-1 nucleocapsid protein in its different maturation states. Viruses. 2020; 12: 1109-28.
- Lavrik IN, Golks A, Krammer PH. Caspases: pharmacological manipulation of cell death. J Clin Invest. 2005; 115: 2665-72.
- Wang X, Tan J, Biswas S, Zhao J, Devadas K, Ye Z, et al. Pandemic influenza A (H1N1) virus infection increases apoptosis and HIV-1 replication in HIV-1 infected Jurkat cells. Viruses. 2016; 8: 33-40.
- Chandrasekar AP, Cummins NM, Badley AD, Chandrasekar AP. The role of the BCL-2 family of proteins in HIV-1 pathogenesis and persistence. Clin Microbiol Rev. 2019; 33: e00107-19.
- Guedán A, Donaldson CD, Caroe ER, Cosnefroy O, Taylor IA, Bishop KN. HIV-1 requires capsid remodelling at the nuclear pore for nuclear entry and integration. PLOS Pathog. 2021; 17: e1009484.
- Martinez-Picado J, Zurakowski R, Buzón MJ, Stevenson M. Episomal HIV-1 DNA and its relationship to other markers of HIV-1 persistence. Retrovirology. 2018; 15: 15.
- Irwan ID, Tax CBR. Induces the recruitment of NF-kappaB to unintegrated HIV-1 DNA to rescue viral gene expression and replication. J Virol. 2021; 95: e0028521.
- Avettand-Fènoël V, Hocqueloux L, Ghosn J, Cheret A, Frange P, Melard A, et al. Total HIV-1 DNA, a marker of viral reservoir dynamics with clinical implications. Clin Microbiol Rev. 2016; 29: 859-80.
- Thorball CW, Borghesi A, Bachmann N, et al. Swiss HIV cohort study. Host Genomics of the HIV-1 Reservoir Size and Its Decay Rate During Suppressive Antiretroviral Treatment. J Acquir Immune Defic Syndr. 2020; 85: 517-24.
- Besson GJ, Lalama CM, Bosch RJ, Gandhi RT, Bedison MA, Aga E, et al. HIV-1 DNA decay dynamics in blood during more than a decade of suppressive antiretroviral therapy. Clin Infect Dis. 2014; 59: 1312-21.