
Research Article
Austin Liver. 2016; 1(1): 1003.
Enzymatic Responses to Alcohol and Tobacco Nicotine- Derived Nitrosamine Ketone Exposures in Long Evans Rat Livers
Yalcin EB¹, Tong M¹ and de la Monte SM1,2*
¹Liver Research Center, Division of Gastroenterology and Department of Medicine, Rhode Island Hospital and the Alpert Medical School of Brown University, USA
²Departments of Neurology, Neurosurgery and Pathology, Rhode Island Hospital and the Alpert Medical School of Brown University, USA
*Corresponding author: de la Monte SM, Departments of Neurology, Neurosurgery and Pathology, Rhode Island Hospital and the Alpert Medical School of Brown University, USA
Received: September 13, 2016; Accepted: November 25, 2016; Published: December 01, 2016
Abstract
Background: Chronic feeding plus binge administration of ethanol causes very high blood alcohol concentrations. However, its co-administration with tobacco Nicotine-Derived Nitrosamine Ketone (NNK) results in somewhat lower blood alcohol levels, suggesting that NNK and therefore smoking, alters alcohol metabolism in the liver. To explore this hypothesis, we examined effects of ethanol and/or NNK exposures on the expression and activity levels of enzymes that regulate their metabolism in liver.
Methods: This study utilized a 4-way model in which Long Evans rats were fed liquid diets containing 0% or 26% ethanol for 8 weeks, and respectively i.p injected with saline or 2 g/kg of ethanol 3 times/week during Weeks 7 and 8. The control and ethanol-exposed groups were each sub-divided and further i.p treated with 2 mg/kg of NNK or saline (3x/week) in Weeks 3-8. ADH, catalase and ALDH activities were measured using commercial kits. CYP450 mRNA levels (17 isoforms) were measured by qRT-PCR analysis.
Results: Ethanol significantly increased hepatic ADH but not catalase or ALDH activity. NNK had no effect on ADH, ALDH, or catalase, but when combined with ethanol, it increased ADH activity above the levels measured in all other groups. Ethanol increased CYP2C7, while NNK increased CYP2B1 and CYP4A1mRNA levels relative to control. In contrast, dual ethanol + NNK exposures inhibited CYP2B1 and CYP4A1 expression relative to NNK. Conclusion: Dual exposures to ethanol and NNK increase hepatic ethanol metabolism, and ethanol and/or NNK exposures alter the expression of CYP450 isoforms that are utilized in NNK and fatty acid metabolism.
Keywords: Alcoholic liver disease; Ethanol metabolism; NNK; Alcohol dehydrogenase; Aldehyde dehydrogenase; Cytochrome P450
Abbreviations
Actβ: Beta-Actin; ADH: Alcohol Dehydrogenase; ALD: Alcoholic Liver Disease; ALDH: Aldehyde Dehydrogenase; ANOVA: Analysis of Variance; CYP: Cytochrome P450; DCA: Dicarboxylic Acid; ER: Endoplasmic Reticulum; HPRT: Hypoxanthine-Guanine Phosphoribosyl transferase; MEOS: Microsomal Ethanol Oxidizing System; NNK: Nicotine Derived Nitrosamine Ketone; qRT-PCR: Quantitative Reverse Transcriptase Polymerase Chain Reaction
Introduction
Severity of alcohol induced liver injury correlates with dose and duration of alcohol exposure and the levels of its toxic metabolites [1,2]. Acute alcohol-mediated liver injury is characterized by reversible steatosis. High levels of chronic or binge alcohol exposures cause simple steatosis to progress through stages including steatohepatitis, fibrosis, and finally cirrhosis [3]. An important mediator of alcoholinduced liver injury is the accumulation of its highly toxic metabolite, acetaldehyde [1]. However, one relatively recent consideration regarding the pathogenesis of progressive alcohol-mediated liver injury is that a very high percentage of heavy drinkers also smoke tobacco products (mainly cigarettes) [4]. Of note is that chronic exposures to ethanol, tobacco smoke/products, and tobacco specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) all cause steatohepatitis with oxidative damage [5-9]. In our efforts to examine the role of NNK as a co-factor in alcoholic liver disease, we made the unexpected observation that dual exposures to NNK and chronic plus binge ethanol administration results in lower blood alcohol concentrations compared with the same ethanol treatments alone [8]. These observations suggested that hepatic enzyme activities needed to metabolize alcohol could be modulated by NNK exposures and therefore probably smoking.
Ethanol is metabolized in the liver through three distinct oxidative pathways including Alcohol Dehydrogenase (ADH), Cytochrome P450 2E1 (CYP2E1), and catalase (Figure 1). ADH is main enzyme responsible for the metabolism of ethanol. ADH is an abundantly expressed NAD+ dependent cytosolic enzyme [10]. The CYP2E1 microsomal ethanol oxidizing pathway requires NADPH as a cofactor [11]. Catalase, localized in peroxisomes, utilizes hydrogen peroxide to catalyze alcohol metabolism [12]. Acetaldehyde, a highly toxic and carcinogenic metabolite is generated by all three oxidative pathways; acetaldehyde can covalently bind and form adducts with DNA [13], phospholipids [14], hepatic microsomal proteins, hemoglobin [15], and erythrocyte membrane proteins [16,17]. Aldehyde Dehydrogenase (ALDH) detoxifies acetaldehyde to acetate in mitochondria [18]. Acetaldehyde accumulations caused by unbalanced increases in ADH or inhibition of ALDH activity lead to increased oxidative and ER stress and cellular injury [19,20]. ADH metabolism of ethanol also yields reduced NADH which promotes steatosis by stimulating fatty acid synthesis [20].
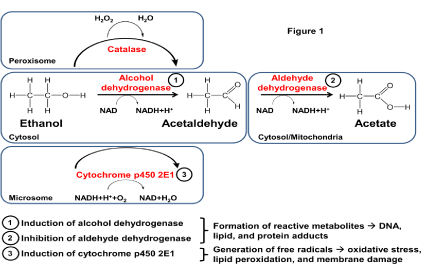
Figure 1: Ethanol Metabolism Pathway.
Ethanol is metabolized in the liver through three distinct oxidative pathways including alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP2E1), and
catalase. ADH, an NAD+ dependent abundant cytosolicenzyme, is responsible for the majority of ethanol metabolism. The second major pathway for alcohol
metabolism is the NADPH cofactor-dependent microsomal ethanol oxidizing system (MEOS) including CYP2E1. The catalase pathway, located in peroxisomes,
is the third key pathway of ethanol oxidation. Each enzymatic pathway produces acetaldehyde, a highly toxic and carcinogenic metabolite that forms adducts with
DNA, lipids, and proteins. Acetaldehyde is detoxified to acetate by aldehyde dehydrogenase (ALDH) in mitochondria. Hepatotoxic effects of ethanol are linked to its
metabolism via ADH, CYP2E1, and ALDH. Induction of ADH or inhibition of ALDH increases hepatic accumulation of acetaldehyde. In addition, CYP2E1 generates
free radicals with attendant oxidative stress, lipid peroxidation, and membrane damage.
NNK is metabolized by reduction of its carbonyl group [21], oxidation of its pyridine nitrogen [22], α-hydroxylation of the methylene carbon adjacent to the N-nitroso nitrogen [23], and α-hydroxylation of methyl carbons adjacent to the N-nitroso nitrogen [24]. Although several CYP450 isoforms can catalyze the α-hydroxylation reactions, CYP2B1 is the dominant isoform expressed in rat liver, whereas CYP1A2, CYP2A1, and CYP3A have minor roles [25,26]. Metabolism of NNK via α-methyl or α-methylene hydroxylation produces reactive metabolites that can methylate [27] or pyridyloxobutylate DNA [28]. In addition, in NNK-associated experimental steatohepatitis, the high hepatic levels of O6 methyl- Guanine adducts may indicate that liver injury is consequential to NNK metabolism [8].
This study was prompted by the finding that dual exposures to ethanol and NNK produced additive adverse histopathological and molecular effects on liver, despite lower blood alcohol levels relative to ethanol-only exposures. One potential explanation for this phenomenon is that NNK increases the rate of ethanol metabolism, yielding higher levels of toxic metabolites and attendant exacerbation of liver injury. Herein, we examined the effects of ethanol, NNK and ethanol + NNK exposures on the mRNA expression or activity levels of enzymes that mediate ethanol and NNK metabolism.
Methods
Experimental model
Liver tissue samples were obtained from a previously generated model [8]. In brief, 4 week old Long Evans male rats were fed isocaloric liquid diets containing 0% or 26% ethanol for 8 weeks (n=12/group). Ethanol-fed rats were binged with 2 g/kg of ethanol by intraperitoneal (i.p.) injection, 3 times per week during the last two weeks of the experiment. Control rats were treated in parallel with i.p. saline. Half the rats in each group were also i.p. injected with NNK (2 mg/kg) or saline 3 times per week from Week 3 through Week 8. Snap-frozen liver tissue harvested at sacrifice and stored at -80°C was used in these studies. The rat experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health, and approved by the Lifespan Institutional Animal Care and Use Committee (IACUC).
Enzyme activity assays
ADH, ALDH, and catalase activities were measured using commercial kits (BioVision, California, USA). Results normalized to sample protein concentration which was measured with the Pierce ™ Thermo Scientific, Rockford, IL) bicinchoninic acid assay. The ADH colorimetric assay is based on alcohol-to-aldehyde interconversion via reduction of NAD+ to NADH and uses isopropanol. ALDH assay is based on oxidation of acetaldehyde, yielding NADH. Changes in absorbance for both the ADH and ALDH assays were measured over 90 minutes at 450 nm. The 10-minute time point was used for inter-group comparisons of enzymatic activity since linear reaction rates were detected within 10 minutes (Figures 2b and 2d). Catalase activity levels were based on the rates in which H2O2 was decomposed to water and oxygen using OxiRed™ to probe unconverted H2O2. Absorbances (570 nm) were inversely proportional to catalase activity.
Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR) Assays
TheRNeasy Mini Kit (Qiagen, Hilden, Germany) was used to extract total RNA from 100 mg samples of fresh frozen liver. RNA was reverse transcribed using the AMV 1st Strand cDNA Synthesis Kit (Roche, Indianapolis, IL). The resulting cDNA templates were used to measure expression levels of 17 cytochrome P450 (CYP) isoforms (Supplementary Table 1) by qPCR in a Roche Lightcycler 480 System as described [29]. Primer pairs were designed using Mac Vector 12 software (Cary,NC). Relative mRNA abundance was calculated using the 2ΔΔCt method with results normalized to Hypoxanthine-guanine phosphoribosyltransferase (HPRT) and Beta-actin (Actβ) as internal controls.
![]()
Enzyme activity
Ethanol Factor
NNK Factor
Ethanol x NNK Interaction
F-Ratio
P-value
F-Ratio
P-Value
F-Ratio
P-Value
ADH
25.32
<0.0001
7.558
0.0124
0.874
N.S.
ALDH
4.939
0.038
0.862
N.S.
0.087
N.S.
Catalase
0.418
N.S.
0.890
N.S.
0.0002
N.S.
Table 1: Ethanol, NNK, and their Interactive Effects on ADH, ALDH, and Catalase Activities. Two-Way ANOVA results from comparing hepatic mean levels of Alcohol dehydrogenase (ADH), aldehyde dehydrogenase (ALDH), and catalase activities in control, ethanol-fed, NNK-exposed and ethanol + NNK treated Long Evans rats (n=6 rats/group).Significant differences are highlighted with bold font. Corresponding data with Tukey post-hoc significance test outcomes are graphed in Figure 2.
Statistical analysis
Results are expressed as mean ± SD. Inter-group comparisons were made by two-way Analysis Of Variance (ANOVA) with Tukey or linear trend post hoc tests (GraphPad Prism 7, San Diego, CA, USA). All assays were performed with 6 independent samples per group since preliminary studies and power analysis determined that this group size was sufficient to achieve 80% power. P-values less than 0.05 were considered as statistically significant.
Results
Effects of ethanol and NNK on alcohol metabolizing enzyme activity levels in liver
Two-way ANOVA tests demonstrated significant ethanol and NNK effects on ADH activity, and significant ethanol effects on ALDH activity (Table 1). In contrast, no significant ethanol, NNK or ethanol x NNK interactive effects were detected for Catalase activity.
ADH activity
Mean (± S.D.) hepatic ADH activity in control (31.7 ± 4.6 nmol/min/mg protein) and NNK-only (38.3 ± 3.5 nmol/min/mg) were similar. Ethanol significantly increased ADH activity (46.5 ± 11.3 nmol/min/mg) by 1.5-fold relative to control (P=0.04). The combined ethanol + NNK treatments increased ADH activity (59.7 ± 12.2 nmol/min/mg) by 1.9-foldrelative to control (P=0.0001) and 1.6-fold relative to NNK exposure (P=0.002) as demonstrated with Tukey post hoc tests (Figure 2a, Table 1).These findings suggest that ethanol increases the metabolic rate for conversion of ethanol to acetaldehyde, and dual ethanol + NNK exposures have additive effects on hepatic ADH activity in rats.
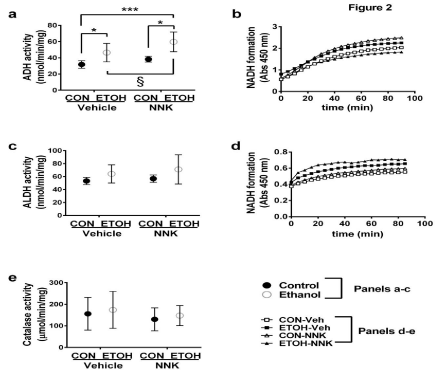
Figure 2: Effects of ethanol and NNK exposures on the ethanol metabolic pathway.
Enzymatic activities of (a,b) alcohol dehydrogenase, (c,d) aldehyde dehydrogenase, and (e) catalase were measured in control, ethanol, NNK, and ethanol + NNK
exposed livers (6/group) by commercially available kits (BioVision, California, USA). Inter-group comparisons were made by two-way ANOVA with the Tukey posttest
(*P< 0.05; **P< 0.01; ***P< 0.001; § 0.05 <P< 0.10).
ALDH activity
The mean levels of hepatic ALDH activity in the control (53.2 ± 5.5 nmol/min/mg) and NNK-exposed (56.8 ± 5.7 nmol/min/mg) groups were similar, whereas the levels in the ethanol (64.1 ± 14.1 n mol/min/mg) and ethanol + NNK(71.0 ± 22.5 nmol/min/mg) were higher. Although significant ethanol effects were observed by two-way ANOVA (Table 2), post-hoc Tukey tests failed to reach statistical significance (Figure 2c, Table 1). Nonetheless, the results demonstrate that ethanol exposure has stimulatory effects on hepatic ALDH activity whereas NNK does not. However, the magnitude of ethanol-induced increase in ALDH activity was modest compared with ADH, suggesting that acetaldehyde metabolism lagged relative to its generation in all 3 experimental groups.
![]()
Gene
Ethanol Factor
NNK Factor
Ethanol x NNK Interaction
F-Ratio
P-Value
F-Ratio
P-Value
F-Ratio
P-Value
CYP1A1
0.087
N.S.
4.72
N.S.
0.103
N.S.
CYP1A2
0.492
N.S.
1.6
N.S.
0.154
N.S.
CYP2A1
0.298
N.S.
2.426
N.S.
0.390
N.S.
CYP2A2
3.831
N.S.
0.0003
N.S.
0.866
N.S.
CYP2B1
15.27
0.005
1.436
N.S.
9.484
0.015
CYP2B2
2.476
N.S.
1.271
N.S.
0.221
N.S.
CYP2C6v1
8.203
0.02
0.006
N.S.
0.0004
N.S.
CYP2C7
6.291
0.037
0.156
N.S.
5.561
0.046
CYP2C11
8.157
0.02
0.468
N.S.
0.105
N.S.
CYP2C12
0.528
N.S.
3.57
N.S.
0.0288
N.S.
CYP2C13
0.543
N.S.
0.198
N.S.
0.0166
N.S.
CYP2D1
0.0006
N.S.
2.081
N.S.
5.93
0.04
CYP2D2
0.5622
N.S.
0.5755
N.S.
6.425
0.035
CYP2E1
14.76
0.005
0.0733
N.S.
0.031
N.S.
CYP3A2
3.043
N.S.
0.003
N.S.
0.075
N.S.
CYP3A23
23.83
0.001
0.019
N.S.
1.912
N.S.
CYP4A1
3.69
N.S.
4.582
N.S.
8.005
0.02
Table 2: Ethanol, NNK, and Their Interactive Effects on Cytochrome P450 mRNA Expression. Two-Way ANOVA results from comparing hepatic mean levels of 17 different cytochrome P450 genes expressed in control, ethanol-fed, NNK-exposed and ethanol + NNK treated Long Evans rats (n=6 rats/group).Gene expression (mRNA) was measured by qPCR analysis. Corresponding data with Tukey multiple comparisons post-hoc significance test outcomes are graphed in Figure 3. Significant differences are highlighted with bold font.
Catalase activity
Hepatic catalase activity, irrespective of treatment, was approximately 1000 times higher than ADH activity. The mean level of catalase activity in control livers(10.1 ± 4.2 μmol/min/mg) was similar to those measured in the ethanol(9.6 ± 3.5 μmol/min/mg), NNK(11.4 ± 4.4 μmol/min/mg), and ethanol + NNK(9.8 ± 3.1 μmol/ min/mg) groups, corresponding with results obtained by two-way ANOVA (Figure 2e, Table 1).
Effects of ethanol and tobacco nitrosamine NNK on hepatic cytochrome P450 expression
Cytochrome P450 (CYP) genes that were measured regulate metabolism of hepatic steroid hormones (CYP1A1, CYP1A2, CYP2A1, CYP2A2, CYP2C11, CYP2C12, CYP2C13, CYP2D1, CYP2E1, CYP3A2, and CYP3A23), fatty acids (CYP1A1, CYP1A2, CYP2B1, CYP2E1, and CYP4A1), and retinoic acid (CYP2C7) and xenobiotics (CYP1A1, CYP1A2, CYP2A1, CYP2C6, CYP2D2, and CYP3A2) or bioactivation of carcinogens (CYP2B1, CYP2B2, and CYP2E1) (Supplementary Table 1). CYP mRNA levels were measured by qRT-PCR analysis.
Two-way ANOVA tests demonstrated significant ethanol effects on the expression levels of CYP2B1, CYP2C6v1, CYP2C7, CYP2C11, CYP2E1, and CYP3A23, and significant ethanol x NNK effects on the expression levels of CYP2B1, CYP2C7, CYP2D1, CYP2D2, and CYP4A1, but no significant NNK effects (Table 2). Post hoc tests demonstrated that ethanol significantly increased expression of CYP2C7, (1.8 fold; P< 0.03) and modestly, but not significantly decreased expression of CYP2C11, CYP2E1, and CYP3A23 relative to control.
NNK increased CYP2B1 expression relative to control (1.9-fold; P< 0.06), ethanol (2.4-fold; P< 0.03), and ethanol + NNK (4.8-fold; P< 0.005) (Figure 3e). In addition, NNK significantly increased CYP4A1 expression relative to control (2.5-fold; P< 0.03) and ethanol + NNK effects (2.5-fold; P=0.04), and had a trend increase relative to ethanolonly effects (1.9-fold; P = 0.08) (Figure 3q). Another notable point was that the mean levels of CYP2E1 (Figure 3n) and CYP3A23 (Figure 3p) were similar in control and NNK livers, whereas ethanol and ethanol + NNK had relative inhibitory effects on the expression of those genes (both P =0.08). Like ethanol, ethanol + NNK exposures inhibited expression of CYP2E1 and CYP3A23 (1.6-fold; P=0.04) relative to control (Figure 2p). In addition, CYP3A23 was also reduced by ethanol + NNK relative to NNK only exposures (1.8-fold; P=0.01).
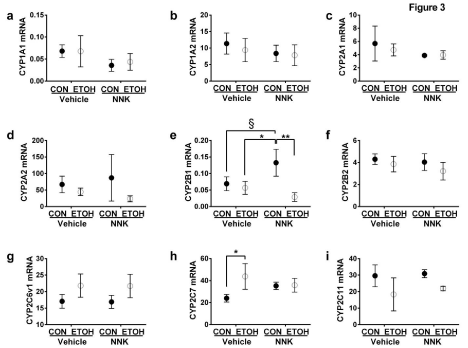
Figure 3: Effects of ethanol and NNK exposures on cytochrome P450 gene expression in Long Evans rat livers.
RNA extracted from control, ethanol, NNK, and ethanol + NNK exposed livers (6/group) was reverse transcribed, and cDNAs were used to measure gene
expression by qPCR analysis. Graphs represent mRNA levels for (a) CYP1A1, (b) CYP1A2, (c) CYP2A1, (d) CYP2A2, (e) CYP2B1, (f) CYP2B2, (g) CYP2C6v1, (h)
CYP2C7, (i) CYP2C11, (j) CYP2C12, (k) CYP2C13, (l) CYP2D1, (m) CYP2D2, (n) CYP2E1, (o) CYP3A2, (p) CYP3A23, and (q) CYP4A1. Inter group comparisons
were made by two-way ANOVA with the Tukey post-test (*P< 0.05; **P< 0.01; ***P< 0.001; § 0.05 <P< 0.10).
Discussion
This study examined the effects of chronic plus binge ethanol, NNK, and ethanol + NNK exposures on the activity or mRNA levels of major enzymes that regulate alcohol and NNK metabolism in liver using a 4-way experimental paradigm generated in adult male Long Evans rats. The research was driven by the earlier observation that dual ethanol + NNK exposures resulted in lower steady-state and post-binge blood alcohol concentrations compared with the same ethanol-only exposures [8].This finding led to the hypothesis that alcohol metabolizing enzymes were modulated by ethanol and/or NNK exposures. This concept is consistent with previously published data demonstrating that experimental chronic ethanol administration increases mRNA, protein, and activity levels of ADH in rat livers [19,30]. Similarly, in other studies, in rats that were infused continuously with ethanol-containing diets, blood and urine ethanol concentrations cycled between 0 and 500 mg/dL [31], but after the urine concentrations reached 300 mg/dL, ADH mRNA expression and activity were induced [32], consistent with adaptive responses to alcohol load. Our studies extend these earlier finding by measuring activity and expression of a broader range of ethanol-metabolizing enzymes, and also consider the effects of independent and coexposures to NNK. The importance of the NNK parallel studies is that consideration is given to the co-factor role of tobacco exposures/ smoking in the pathogenesis of alcohol-mediated liver injury.
In this study, we demonstrated that ethanol, ethanol + NNK significantly increase ADH but just modestly increase ALDH relative to control, with larger effect sizes produced by the dual exposures. Although NNK alone had no detectable effect on ADH or ALDH activity relative to control, the finding that ethanol + NNK had somewhat additive responses compared with ethanolalone suggests that NNK, and therefore probably also smoking, has modifying effects on ethanol-stimulated ADH and ALDH activities. The greater increase in ADH activity associated with ethanol + NNK versus ethanol-only exposures accounts for the lower blood alcohol concentrations measured in the dual-exposure group. On the other hand, the disproportionately greater increases in ADH compared with ALDH activity effectuated by both ethanol and ethanol + NNK favored excess formation and accumulation of acetaldehyde in liver. In addition, the larger effects of ethanol + NNK versus ethanol suggest that hepaticacetaldehyde levels were higher in the dual exposure group, an effect that could account for the greater severity of liver injury compared with the ethanol-only group.
Mechanistically, acetaldehyde’s highly toxic and carcinogenic [17] effects are due to its strong electrophilic structure and ability to form adducts with nucleophilic cellular components (DNA, RNA, and protein), leading to diverse pathophysiological responses including enzyme inactivation, DNA damage, and increased cell death [33]. Impairment of cellular enzyme functions leads to increased formation of reactive metabolites and free radicals that promote oxidative stress, lipid peroxidation, and membrane damage, exacerbating tissue injury and organ dysfunction. Therefore, detoxification of acetaldehyde via ALDH activation is a vital cellular protective mechanism that has therapeutic implications [33,34]. In regard to the present work, it is likely that acetaldehyde accumulations mediated by the minimal or absent activation of ALDH vis-à -vis significant increases in ADH activity significantly contributed to liver injury following ethanol and/or NNK exposures.
Among the 17 CYP isoforms included in the qPCR array, the expression levels of 9 (53%) were significantly modulated by ethanol (n=6) and/or ethanol + NNK (n=5) exposures. Just two of those CYP genes were significantly altered by both ethanol and ethanol + NNK, and NNK-only effects were not observed. These findings suggest that NNK and therefore smoking, differentially modifies hepatic profiles of CYP gene expression in the context of chronic binge ethanol exposures.
NNK is primarily oxidized by CYP2B1 [35], and less commonly by CYP1A2 and CYP2A1 in rat livers [25,26]. Induction of CYP2B1 activates NNK via α-methyl or α-methylene hydroxylation pathways. Hydroxylated NNK metabolites can form methyl or pyridyloxobutyl adducts with DNA [27,28]. Therefore, the NNK associated increases in CYP2B1 expression may have contributed to liver injury due to accumulation of DNA adducts and attendant DNA damage.
Previous studies showed that ethanol induces hepatic CYP2E1 protein levels and enzyme activity [36], and it impairs mitochondrial β-oxidation but stimulates ω-oxidation of free fatty acids in reactions catalyzed by microsomal CYPs, including mainly by CYP4A1 and CYP2E1 [37]. However, in the present study, ethanol and ethanol + NNK exposures resulted in modest reductions in CYP2E1, and no change in CYP4A1 mRNA levels. Discrepancies between mRNA and protein expression or enzyme activity occur under various conditions. One potential explanation for the reduced mRNA levels is adduct formation mediated by metabolites such as acetaldehyde. In contrast, NNK exposed rat livers had significant or trend elevations in CYP4A1 expression relative to the control, ethanol and ethanol + NNK groups. Increased levels of ω-oxidation of long chain fatty acids produce toxic Dicarboxylic Acids (DCAs) and ω-hydroxylated fatty acids, which impair mitochondrial oxidative phosphorylation and promote steatohepatitis [37,38]. Therefore, NNK’s selective stimulatory effects on CYP4A1 may have been an important mediator of steatohepatitis in that group.
CYP2C7, a constitutive enzyme involved in the oxidative metabolism of vitamin A (retinol) and retinoic acid [39,40], was previously shown to be stimulated by ethanol in rat liver microsomes [36]. Correspondingly, hepatic CYP2C7 expression was also significantly increased by ethanol and exhibited statistical trend elevations in the ethanol + NNK group. A potential consequence of CYP2C7 induction by ethanol is that with the increased catabolism of retinoic acid and attendant elimination of retinol (Vitamin A) from the body, hepatic stores of vitamin A could be rendered deficient, which occurs commonly in alcoholics [41,42].
In conclusion, the findings of this study suggest that chronic alcohol and NNK exposures significantly alter hepatic expression, activation and profiles of enzymes used to metabolize ethanol and/ or NNK. These effects of ethanol and/or NNK may contribute to the pathogenesis of steatohepatitis by increasing hepatic accumulation of ethanol and NNK mediated DNA adducts. These data provide a better understanding of the mechanisms of alcohol-induced liver injury and the contribution of tobacco NNK or smoking as a cofactor in the pathogenesis of alcoholic liver disease.
Acknowledgement
The research was supported by grants F32 AA024018-01 (EBY) and AA-11431 (SMdlM) from the National Institutes on Alcohol Abuse and Alcoholism at the National Institutes of Health.
References
- Massey VL, Arteel GE. Acute alcohol-induced liver injury. Frontiers in physiology. 2012; 3: 193.
- Seth D, Haber PS, Syn WK, Diehl AM, Day CP. Pathogenesis of alcohol-induced liver disease: classical concepts and recent advances. Journal of gastroenterology and hepatology. 2011; 26: 1089-1105.
- O'Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. The American journal of gastroenterology. 2010; 105: 14-32.
- Romberger DJ, Grant K. Alcohol consumption and smoking status: the role of smoking cessation. Biomed Pharmacother. 2004; 58: 77-83.
- Van Skike CE, Maggio SE, Reynolds AR, Casey EM, Bardo MT, Dwoskin LP, et al. Critical needs in drug discovery for cessation of alcohol and nicotine polysubstance abuse. Progress in neuro-psychopharmacology & biological psychiatry. 2016; 65: 269-287.
- Zein CO. Clearing the smoke in chronic liver diseases. Hepatology. 2010; 51: 1487-1490.
- Azzalini L, Ferrer E, Ramalho LN, Moreno M, Dominguez M, Colmenero J, et al. Cigarette smoking exacerbates nonalcoholic fatty liver disease in obese rats. Hepatology. 2010; 51: 1567-1576.
- Zabala V, Tong M, Yu R, Ramirez T, Yalcin EB, Balbo S, et al. Potential contributions of the tobacco nicotine-derived nitrosamine ketone (NNK) in the pathogenesis of steatohepatitis in a chronic plus binge rat model of alcoholic liver disease. Alcohol and alcoholism. 2015; 50: 118-131.
- de la Monte SM, Tong M, Agawal AR, Cadenas E. Tobacco Smoke-Induced Hepatic Injury with Steatosis, Inflammation, and Impairments in Insulin and Insulin-Like Growth Factor Signaling. J Clin Exp Pathol. 2016; 6: 269.
- Lieber CS, DeCarli LM, Feinman L, Hasumura Y, Korsten M, Matsuzaki S, et al. Effect of chronic alcohol consumption on ethanol and acetaldehyde metabolism. Advances in experimental medicine and biology. 1975; 59: 185-227.
- Lieber CS, Rubin E, DeCarli LM. Hepatic microsomal ethanol oxidizing system (MEOS): Differentiation from alcohol dehydrogenase and NADPH oxidase. Biochemical and biophysical research communications. 1970; 40: 858-865.
- Keilin D, Hartree EF. Properties of catalase. Catalysis of coupled oxidation of alcohols. The Biochemical journal. 1945; 39: 293-301.
- Balbo S, Hashibe M, Gundy S, Brennan P, Canova C, Simonato L, et al. N2-ethyldeoxyguanosine as a potential biomarker for assessing effects of alcohol consumption on DNA. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008; 17: 3026-3032.
- Kenney WC. Formation of Schiff base adduct between acetaldehyde and rat liver microsomal phosphatidylethanolamine. Alcoholism, clinical and experimental research. 1984; 8: 551-555.
- Lin RC, Lumeng L. Formation of the 37KD protein-acetaldehyde adduct in liver during alcohol treatment is dependent on alcohol dehydrogenase activity. Alcoholism, clinical and experimental research. 1990; 14: 766-770.
- Niemela O, Parkkila S. Alcoholic macrocytosis--is there a role for acetaldehyde and adducts? Addiction biology. 2004; 9: 3-10.
- Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxidative medicine and cellular longevity. 2010; 3: 178-185.
- Eriksson CJ. Acetaldehyde metabolism in vivo during ethanol oxidation. Advances in experimental medicine and biology. 1977; 85: 319-341.
- Ronis MJ, Korourian S, Blackburn ML, Badeaux J, Badger ™. The role of ethanol metabolism in development of alcoholic steatohepatitis in the rat. Alcohol. 2010; 44: 157-169.
- Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004; 34: 9-19.
- Hecht SS, Spratt TE, Trushin N. Absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 1997; 18: 1851-1854.
- Staretz ME, Murphy SE, Patten CJ, Nunes MG, Koehl W, Amin S, et al. Comparative metabolism of the tobacco-related carcinogens benzo[a]pyrene, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol, and N'- nitrosonornicotine in human hepatic microsomes. Drug metabolism and disposition: the biological fate of chemicals. 1997; 25: 154-162.
- Peterson LA, Mathew R, Hecht SS. Quantitation of microsomal alpha-hydroxylation of the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer research. 1991; 51: 5495-5500.
- Murphy SE, Spina DA, Nunes MG, Pullo DA. Glucuronidation of 4-((hydroxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone, a metabolically activated form of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, by phenobarbital-treated rats. Chemical research in toxicology. 1995; 8: 772-779.
- Guo Z, Smith TJ, Ishizaki H, Yang CS. Metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) by cytochrome P450IIB1 in a reconstituted system. Carcinogenesis. 1991; 12: 2277-2282.
- Guo Z, Smith TJ, Thomas PE, Yang CS. Metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by inducible and constitutive cytochrome P450 enzymes in rats. Archives of biochemistry and biophysics. 1992; 298: 279-286.
- Peterson LA, Carmella SG, Hecht SS. Investigations of metabolic precursors to hemoglobin and DNA adducts of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 1990; 11: 1329-1333.
- Peterson LA, Mathew R, SE BPM, Trushin N, Hecht SS. In vivo and in vitro persistence of pyridyloxobutyl DNA adducts from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 1991; 12: 2069-2072.
- Tong M, Yu R, Silbermann E, Zabala V, Deochand C, de la Monte SM. Differential Contributions of Alcohol and Nicotine-Derived Nitrosamine Ketone (NNK) to White Matter Pathology in the Adolescent Rat Brain. Alcohol and alcoholism. 2015; 50: 680-689.
- He L, Ronis MJ, Badger ™. Ethanol induction of class I alcohol dehydrogenase expression in the rat occurs through alterations in CCAAT/enhancer binding proteins beta and gamma. The Journal of biological chemistry. 2002; 277: 43572-43577.
- Tsukamoto H, French SW, Reidelberger RD, Largman C. Cyclical pattern of blood alcohol levels during continuous intragastric ethanol infusion in rats. Alcoholism, clinical and experimental research. 1985; 9: 31-37.
- Badger TM, Hoog JO, Svensson S, McGehee RE, Jr., Fang C, Ronis MJ, et al. Cyclic expression of class I alcohol dehydrogenase in male rats treated with ethanol. Biochemical and biophysical research communications. 2000; 274: 684-688.
- Dolle L, Gao B. Pharmacological chaperone therapies: Can aldehyde dehydrogenase activator make us healthier? Journal of hepatology. 2015; 62: 1228-1230.
- Zhong W, Zhang W, Li Q, Xie G, Sun Q, Sun X, et al. Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol-induced hepatic steatosis and cell death in mice. Journal of hepatology. 2015; 62: 1375-1381.
- Yue J, Khokhar J, Miksys S, Tyndale RF. Differential induction of ethanol-metabolizing CYP2E1 and nicotine-metabolizing CYP2B1/2 in rat liver by chronic nicotine treatment and voluntary ethanol intake. European journal of pharmacology. 2009; 609: 88-95.
- Hakkak R, Korourian S, Ronis MJ, Ingelman-Sundberg M, Badger TM. Effects of diet and ethanol on the expression and localization of cytochromes P450 2E1 and P450 2C7 in the colon of male rats. Biochemical pharmacology. 1996; 51: 61-69.
- Hardwick JP, Osei-Hyiaman D, Wiland H, Abdelmegeed MA, Song BJ. PPAR/RXR Regulation of Fatty Acid Metabolism and Fatty Acid omega-Hydroxylase (CYP4) Isozymes: Implications for Prevention of Lipotoxicity in Fatty Liver Disease. PPAR research. 2009; 2009: 52734.
- Grasselli E, Voci A, Demori I, De Matteis R, Compalati AD, Gallo G, et al. Effects of binge ethanol on lipid homeostasis and oxidative stress in a rat model of nonalcoholic fatty liver disease. Journal of physiology and biochemistry. 2014; 70: 341-353.
- Leo MA, Lieber CS. New pathway for retinol metabolism in liver microsomes. The Journal of biological chemistry. 1985; 260: 5228-5231.
- Leo MA, Iida S, Lieber CS. Retinoic acid metabolism by a system reconstituted with cytochrome P-450. Archives of biochemistry and biophysics. 1984; 234: 305-312.
- Clugston RD, Blaner WS. The adverse effects of alcohol on vitamin A metabolism. Nutrients. 2012; 4: 356-371.
- Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol research & health: the journal of the National Institute on Alcohol Abuse and Alcoholism. 2003; 27: 220-231.