
Review Article
Austin Med Sci. 2016; 1(1): 1001.
22q11.2 Deletion Syndrome: Unmasking the Role of Tbx1 in Craniofacial Development
Funato N*
Human Gene Sciences Center, Tokyo Medical and Dental University, Japan
*Corresponding author: Funato N, Research Center for Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8510, Japan
Received: December 18, 2015; Accepted: January 19, 2016; Published: January 21, 2016
Abstract
T-box transcription factor gene (TBX1) is thought to be responsible for chromosome 22q11.2 deletion syndrome (DiGeorge/velocardiofacial syndrome), which is characterized by craniofacial defects, cardiac malformations, thymic and parathyroid hypoplasia, and cleft palates. TBX1 regulates the cell fate of progenitor cells in cranial and pharyngeal tissues during embryogenesis. In this review, I discuss the mechanisms of TBX1 during craniofacial development of tissues including the palate, bones, teeth, and muscle.
Keywords: DiGeorge syndrome; Velocardiofacial syndrome; Pharyngeal arch; Palatogenesis; Bone anomalies; Cleidocranial dysplasia
Abbreviations
22q11.2DS: 22q11.2 deletion syndrome; CP: cleft palate; SNP: single nucleotide polymorphism; CCD: cleidocranial dysplasia; CL: cervical loop; FGF: fibroblast growth factor
Introduction
TBX1, a member of the T-box transcription factor gene family, is considered to be a candidate gene for chromosome 22q11.2 deletion syndrome (22q11.2DS). 22q11.2DSmanifests as DiGeorge syndrome (OMIM 188400), velocardiofacial syndrome (OMIM 192430), and conotruncal anomaly face syndrome (OMIM 217095).22q11.2DS is the most frequent micro-deletion syndrome, affecting approximately 1 in 4000 live births, and is characterized by a series of phenotypic abnormalities, including craniofacial anomalies, cardiovascular defects, thymic and parathyroid hypoplasia, velopharyngeal insufficiency, and skeletal muscle hypotonia [1-4]. Structures primarily affected in 22q11.2DS are derivatives of the pharyngeal arches and head mesenchyme [4,5].
TBX1 is expressed in the pharyngeal tissues, including mesoderm, ectoderm, and endoderm, and throughout the head mesenchyme in mice [6-8] (Figure 1A). TBX1knockout (Tbx1–/–) mice exhibit most features of 22q11.2DS, including cardiac, craniofacial, thymic, and parathyroid defects, and skeletal muscle hypotonia [1-3,8,9]. Craniofacial anomalies occur in ~60% of 22q11.2DS patients [5]; the most frequent craniofacial defects include micrognathia, ear abnormalities, hypertelorism, blunted nose, various degrees of Cleft Palate (CP), and tooth defects [5,10,11]. This review focuses on the functions of TBX1incraniofacial development.
Tbx1 and cleft palate
CP is the most frequent craniofacial birth defects in humans, occurring in 1 in 500 to 1000 live births worldwide [6]. The craniofacial malformations observed in 22q11DS patients include various subtypes of CP (complete CP, incomplete CP, sub mucosal CP, and bifid uvula). TBX1 mutations have been identified in patients with characteristic phenotypes of 22q11.2DS, conotruncal anomaly face syndrome (OMIM 217095), and nonsyndromic CP [12,13]. These findings suggest that TBX1 is a potential candidate gene for various degrees of nonsyndromic CP. Indeed, two adjacent Single Nucleotide Polymorphisms (SNPs) upstream of TBX1 suggest a potential association with the CP phenotype, although they are not significant after correcting for multiple testing [14]. Regulatory elements for TBX1 expression on 22q11.2 may also be involved in the CP phenotype.
TBX1 is expressed in both the anterior and posterior edges of the paired palatal shelves in mice, highlighting the intrinsic function of TBX1 in regulating palatogenesis. Deletion of TBX1 results in abnormal epithelial fusion between the palatal shelves and the mandible, which induces CP by inhibiting elevation of the palatal shelves [15]. Tbx1–/– mice present various degrees of CP phenotype, including complete CP, incomplete CP, submucosal CP, and anterior CP, whereas Tbx1+/–mice are phenotypically normal [15] (Table 1). Ablation of TBX1 specifically in the epithelial cells (TBX1LoxP/KO; KRT14-Cre) results in anterior CP [15]. The variations in palatal phenotypes across different TBX1 mutants strongly suggest that TBX1 is involved in stochastic factors and/or modifier genes. Expression of Pax9, mutations of which lead to CP and adontogenesis [16], is down-regulated in the pharyngeal arch and palatal shelves of Tbx1- /- embryos [15,17]. Tbx1–/– hyperproliferative epithelium displays incomplete differentiation, suggesting that TBX1 controls the balance between proliferation and differentiation of epithelial cells. CP phenotypes of Tbx1–/– mice suggest that the various degrees of CP phenotypes could be induced by the pathogenic adhesion-separation of the oral epithelium, together with compromised growth of palatal mesenchyme [15,18].
![]()
Gene
Genetic Disease/Mice
Palatal phenotype
Skeletal phenotype
TBX1
22q11.2DS
(OMIM 188400, 192430, 217095, 217095)
CP, submucosal CP, bifid uvula
Short stature, microcephaly, retrognathia, micrognathia, blunted nose, hypoplastic nasal alae
RUNX2
CCD syndrome
(OMIM 119600)
Submucosal CP
CCD syndrome*
Tbx1
Tbx1+⁄−mice
Normal
Normal
Tbx1−⁄− mice
CP, incomplete CP, submucosal CP [15]
Normal
Tbx1LoxP/KO; KRT14-Cre mice (epithelium)
Anterior CP [15]
Normal [8]
Tbx1LoxP/KO; Mesp1-Cre mice (mesoderm)
Normal
CCD syndrome-like*, hypomorphic HB [8]
Tbx1LoxP/KO; Twist2-Cre mice (bone primordium)
Normal
CCD syndrome-like*, no mineralized HB [8]
Tbx1LoxP/KO; Wnt1-Cre mice (neural crest)
Normal
No mineralized HB[8]
Runx2
Runx2+⁄−mice
Not reported
CCD syndrome*, HB hypoplasia [25]
Runx2−⁄− mice
CP [37]
No mineralized bone [23,25]
22q11.2DS: 22q11.2 Deletion Syndrome; CCD: Cleidocranial Dysplasia; CP: Cleft Palate; HB: Hyoid Bone *hypoplastic clavicle, abnormal neurocranium morphology, and a nasal bone defect
Table 1: Skeletal and palatal phenotypes of human genetic disease and mouse mutants.
Tbx1 and bone abnormalities
22q11.2DS patients manifest with craniofacial malformations including short stature, brachycephaly, micrognathia, blunted nose, hypertelorism, and small low-set ears [3,19]. Tbx1–/–mice also display skeletal abnormalities, such as short stature, persistently open fontanelles, micrognathia, hypoplasia of clavicle and zygomatic arch, small low-set ears, and absence of hyoid bone [8] (Figure 1B). A cell type-specific deletion of TBX1 in the mesoderm (TBX1LoxP/KO; Mesp1- Cre) or osteochondral progenitors (TBX1LoxP/KO; Twist2-Cre) partially recapitulates the Tbx1–/–bone phenotypes (Table 1). TBX1 is involved in the following aspects of osteogenesis. First, TBX1 is expressed in mesoderm-derived calvarial bone primordium and is directly involved in calvarial bone development. Loss of TBX1 in the cranial mesoderm (TBX1LoxP/KO; Mesp1-Cre) or osteochondral progenitors (TBX1LoxP/KO; Twist2-Cre) impairs development of TBX1-progeny calvarial bones [8]. Second, TBX1 expression in neural crest cells or in osteochondral progenitors is necessary for the morphogenesis and ossification of the hyoid bone. Interestingly, 22q11.2DSpatients exhibit delayed development of the hyoid bone [20], ordain visible hyoid ossification center [21]. Abnormalities in other neural crest-derived bones, such as frontal bones, mandibular bones, and temporal bones are secondary defects induced by non-neural crest cells in Tbx1–/–mice [8,22]. It is likely that TBX1 expression in the cranial endoderm and mesoderm affects development of adjacent neural crest derivatives. Tbx1–/–mice also have bone abnormalities in the endochondral bones, including the atlas, axis, xiphoid process, and the cranial base. The synchondroses in the Tbx1–/–cranial base are completely ossified [3,8,9]. These results indicate that TBX1 is required for mesodermand neural crest-derived bone morphogenesis and ossification.
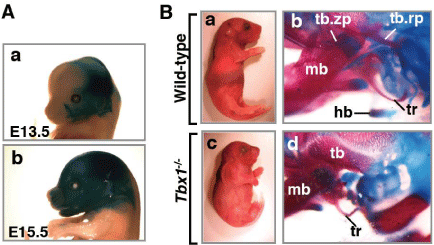
Figure 1: Craniofacial malformations in Tbx1mutants.
(A) Lateral view of the head of whole-mount lacZ-stained Tbx1-Cre; R26R
mice at E13.5 (a) and E15.5 (b). LacZ-positive cells are widely detectable in
the head. (B) Craniofacial phenotype of Tbx1-/- mice. (a,c) Appearance of P1
wild-type (a) and Tbx1-/- (c) mice. Tbx1-/- mutants (c) are small compared to
wild-type mice (a).(b,d) Alizarin red (for mineralized bone) and alcian blue (for
cartilage) staining of wild-type (b) and Tbx1-/- (d) bones of the neck. In Tbx1-/- mice, the hyoid bone is a plastic, and the tympanic ring and the processes
of the temporal bone are hypoplastic (d). hb, hyoid bone; tr, tympanic ring;
tb, temporal bone; tb.rp, retroarticular process of temporal bone; tb.zp,
zygomatic process of temporal bone; mb, mandibular bone.
All animal experimental procedures were reviewed and approved by the
Institutional Animal Care and Use Committee of the Tokyo Medical and
Dental University and University of Texas Southwestern Medical Center.
Skeletal abnormalities observed in Tbx1–/–mice are similar to those of cleidocranial dysplasia (CCD, OMIM 119600) in humans, which is caused by heterozygous mutation in the Runx2 gene (Table 1). Runx2, a member of the Runt-related transcription factors, is essential for osteoblast differentiation, and Runx2–/–mice display no mineralized bones [23-25]. Similar to CCD in humans, Runx2+⁄− mice exhibit short stature, abnormal neurocranium morphology, persistently open fontanel’s, nasal bone defects, and hypoplasia of the clavicle, hyoid bone, and zygomatic arch [23,25]. Deletion of TBX1affects Runx2 expression in parietal bones, suggesting that TBX1 may be involved in the maintenance of cell populations expressing Runx2at the onset of bone development. Since TBX1overexpression induces Runx2 expression in vitro, it is also possible that TBX1 may act upstream of Runx2. These results suggest that TBX1 mutations could lead to CCD-like skeletal phenotypes in humans and TBX1 may be the candidate gene for recessive inheritance of CCD (OMIM 216330).
Tbx1 and dental anomalies
In 22q11DS patients, dental anomalies (enamel hypoplasia, hypomineralization, single central incisor, and small teeth) have been reported [10,11]. Similar to the human phenotype, the upper incisors are absent in 30% of Tbx1–/–mice [15]. In developing teeth, Tbx1 expression is controlled by Fibroblast Growth Factor (FGF) signaling [26]. Mouse incisors have the dental stem cell niche in the labial and lingual Cervical Loops (CLs).SinceTbx1 is expressed in the CLs, Tbx1 may regulate the stem cell niche. Inactivation of Tbx1 specifically in the keratinocyte lineage results in a slightly smaller tooth compared to wild-type [27]. The CL region of the incisoris either severely reduced or completely missing in Tbx1–/–incisors [28]. Tbx1 binds to Pitx2 (paired-like homeodomain transcription factor 2) and activates promoters of Pitx2and Cdkn1a (a cyclin-dependent kinase inhibitor 1A, also known as p21), suggesting that Tbx1 regulates cell proliferation in the dental epithelium through the Pitx2-Cdkn1a axis [29]. Tbx1 regulates the transition between stem cell quiescence and proliferation in hair follicles [30]. Tbx1 in the CLsmay also regulate the proliferation, differentiation, and/or maintenance of the stem cell niche.
Tbx1 and Muscle Hypotonia
The branchiomeric muscles are derived from the mesodermal core of the pharyngeal arches [31]. In Tbx1–/–embryos, branchiomeric muscles, including the masseter, pterygoid, and temporalis muscles, are intermittently absent [32], suggesting that Tbx1 is required for determining cell fate and survival of the myogenic cells in branchiomeric muscles. In branchiomeric muscle formation, the basic helix–loop–helix transcriptionfactorsTcf21 (transcription factor 21, also known as capsulin), Msc (musculin, also known as MyoR), Myf5 (myogenic factor 5), and Myod1 (myogenic differentiation 1), and other transcription factors such as Pitx2 and Isl1 (ISL1 transcription factor, LIM/homeodomain) play critical roles [33-35]. Tbx1 functions downstream of Tcf21, Pitx2, and Isl1, and upstream of Lhx2 (LIM homeobox protein 2), Myf5, Myod1, Tlx1 (T cell leukemia, homeobox 1), and Fgf10 in cardiogenesis or myogenesis [32-34,36]. Therefore, Tbx1 controls the onset and the development of branchiomeric muscles through transcriptional regulation of myogenic determination genes.
Conclusion
Progress has been made to establish how Tbx1 contributes to the palatal, craniofacial, dental, and muscle phenotypes observed in 22q11.2DS patients. Epigenetics and the microRNA regulation could change Tbx1 expression [27,38]. Further research is needed to elucidate the genetic, cellular, and molecular roles of Tbx1 and apply this knowledge to the management of 22q11.2DS patients in the future.
Acknowledgement
I thank for Deepack Srivastava for mice and Hiromi Yanagisawa for critical reading of the manuscript. This work was supported by JSPS KAKENHI Grant Numbers 25670774 and15K11004.
References
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001; 410: 97-101.
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001; 104: 619-629.
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001; 27: 286-291.
- Scambler PJ. The 22q11 deletion syndromes. Hum Mol Genet. 2000; 9: 2421-2426.
- Gorlin RJ, Cohen MM, Hennecam RCM. Syndromes of the head and the neck. 4th edn. New York; Oxford University Press. 2001.
- Tolarová MM, Cervenka J. Classification and birth prevalence of orofacial clefts. Am J Med Genet. 1998; 75: 126-137.
- Jones MC. Etiology of facial clefts: prospective evaluation of 428 patients. Cleft Palate J. 1988; 25: 16-20.
- Funato N, Nakamura M, Richardson JA, Srivastava D, Yanagisawa H. Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Hum Mol Genet. 2015; 24: 424-435.
- Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing auto regulatory loop involving fork head transcription factors. Development. 2004; 131: 5491-5502.
- Oberoi S, Vargervik K. Velocardiofacial syndrome with single central incisor. Am J Med Genet A. 2005; 132A: 194-197.
- Klingberg G, Oskarsdóttir S, Johannesson EL, Norén JG. Oral manifestations in 22q11 deletion syndrome. Int J Paediatr Dent. 2002; 12: 14-23.
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003; 362: 1366-1373.
- Osoegawa K, Vessere GM, Utami KH, Mansilla MA, Johnson MK, Riley BM, et al. Identification of novel candidate genes associated with cleft lip and palate using array comparative genomic hybridization. J Med Genet. 2008; 45: 81-86.
- Herman SB, Guo T, McGinn DM, Blonska A, Shanske AL, Bassett AS, et al. Overt cleft palate phenotype and TBX1 genotype correlations in velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Am J Med Genet A. 2012; 158A: 2781-2787.
- Funato N, Nakamura M, Richardson JA, Srivastava D, Yanagisawa H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum Mol Genet. 2012; 21: 2524-2537.
- Peters H, Neubüser A, Kratochwil K, Balling R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998; 12: 2735-2747.
- Ivins S, Lammerts van Beuren K, Roberts C, James C, Lindsay E, Baldini A, et al. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. DevBiol. 2005; 285: 554-569.
- Goudy S, Law A, Sanchez G, Baldwin HS, Brown C. Tbx1 is necessary for palatal elongation and elevation. Mech Dev. 2010; 127: 292-300.
- Yamagishi H, Maeda J, Hu T, McAnally J, Conway SJ, Kume T, Kume T, et al. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 2003; 17: 269-281.
- Heliövaara A, Ranta R, Rautio J. Pharyngeal morphology in children with submucous cleft palate with and without surgery. Eur Arch Otorhinolaryngol. 2005; 262: 534-538.
- Wells TR, Gilsanz V, Senac MO Jr, Landing BH, Vachon L, Takahashi M. Ossification centre of the hyoid bone in DiGeorge syndrome and tetralogy of Fallot. Br J Radiol. 1986; 59: 1065-1068.
- Aggarwal VS, Carpenter C, Freyer L, Liao J, Petti M, Morrow BE. Mesodermal Tbx1 is required for patterning the proximal mandible in mice. Dev Biol. 2010; 344: 669-681.
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997; 89: 755-764.
- Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004; 18: 952-963.
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997; 89: 765-771.
- Mitsiadis TA, Tucker AS, De Bari C, Cobourne MT, Rice DP. A regulatory relationship between Tbx1 and FGF signaling during tooth morphogenesis and ameloblast lineage determination. Dev Biol. 2008; 320: 39-48.
- Gao S, Moreno M, Eliason S, Cao H, Li X, Yu W, et al. TBX1 protein interactions and microRNA-96-5p regulation controls cell proliferation during craniofacial and dental development: implications for 22q11.2 deletion syndrome. Hum Mol Genet. 2015; 24: 2330-2348.
- Catón J, Luder HU, Zoupa M, Bradman M, Bluteau G, Tucker AS, et al. Enamel-free teeth: Tbx1 deletion affects amelogenesis in rodent incisors. Dev Biol. 2009; 328: 493-505.
- Cao H, Florez S, Amen M, Huynh T, Skobe Z, Baldini A, et al. Tbx1 regulates progenitor cell proliferation in the dental epithelium by modulating Pitx2 activation of p21. Dev Biol. 2010; 347: 289-300.
- Chen T, Heller E, Beronja S, Oshimori N, Stokes N, Fuchs E. An RNA interference screen uncovers a new molecule in stem cell self-renewal and long-term regeneration. Nature. 2012; 485: 104-108.
- Trainor PA, Tan SS, Tam PP. Cranial paraxial mesoderm: regionalisation of cell fate and impact on craniofacial development in mouse embryos. Development. 1994; 120: 2397-2408.
- Kelly RG, Jerome-Majewska LA, Papaioannou VE. The del22q11.2 candidate gene Tbx1 regulates branchiomeric myogenesis. Hum Mol Genet. 2004; 13: 2829-2840.
- Dong F, Sun X, Liu W, Ai D, Klysik E, Lu MF, et al. Pitx2 promotes development of splanchnic mesoderm-derived branchiomeric muscle. Development. 2006; 133: 4891-4899.
- Harel I, Maezawa Y, Avraham R, Rinon A, Ma HY, Cross JW, et al. Pharyngeal mesoderm regulatory network controls cardiac and head muscle morphogenesis. Proc Natl Acad Sci USA. 2012; 109: 18839-18844.
- Nathan E, Monovich A, Tirosh-Finkel L, Harrelson Z, Rousso T, Rinon A, et al. The contribution of Islet1-expressing splanchnic mesoderm cells to distinct branchiomeric muscles reveals significant heterogeneity in head muscle development. Development. 2008; 135: 647-657.
- Grifone R, Jarry T, Dandonneau M, Grenier J, Duprez D, Kelly RG. Properties of branchiomeric and somite-derived muscle development in Tbx1 mutant embryos. Dev Dyn. 2008; 237: 3071-3078.
- Aberg T, Cavender A, Gaikwad JS, Bronckers AL, Wang X, Waltimo-Sirén J, et al. Phenotypic changes in dentition of Runx2 homozygote-null mutant mice. J Histochem Cytochem. 2004; 52: 131-139.
- Wang J, Bai Y, Li H, Greene SB, Klysik E, Yu W, et al. MicroRNA-17-92, a direct Ap-2α transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet. 2013; 9: e1003785.