
Special Article - Neurology
Austin Med Sci. 2018; 3(1): 1020.
The Great Mimicker Carbon Monoxide in Neurology
Dubey A1* and Dubey S2
1Department of Neurology, SAIMS Medical College and PG Institute, India
2Department of Medicine, Gandhi Medical College and Hamidia Hospital, India
*Corresponding author: Ayush Dubey, Department of Neurology, SAIMS Medical College and PG Institute, Indore, India
Received: December 04, 2017; Accepted: January 24, 2018; Published: January 31, 2018
Abstract
Carbon Monoxide is an established acute as well as chronic toxicant which arises from incomplete combustion and occurs in a variety of occupational and environmental situations. It has been known to cause various symptoms including severe neurological ones too. It has got a complex and varied mechanism of cellular toxicity. Delayed neurological deficits are less recognized and often not attributed to the toxicity. Radiological findings are often less specific and sometimes reversible. A comprehensive review on this important yet relatively less reported entity is discussed.
Keywords: Carbon Monoxide; Hyperbaric oxygen therapy
Abbreviations
CO: Carbon Monoxide; HBOT: Hyper Baric Oxygen Therapy; NBOT: Normo Baric Oxygen Therapy; CO-Hgb: Carboxyhaemoglobin
Introduction
Carbon Monoxide (CO) is a colorless, odorless, non irritating gas produced by incomplete burning of carbon containing fossil fuels [1]. It is the leading cause of poisoning related mortality in the United States and is responsible for more than half of all the fatal poisonings occurring worldwide.
CO is often referred to as a “great mimicker” or “great imitator” because the clinical presentations associated with its toxicity are non specific and diverse including headache, chest pain, syncope, seizures and flu like illness. Undiagnosed exposure may often lead to a significant morbidity and mortality [2,3].
Environmental CO exposure is usually less than 0.001% or around 10 ppm [4], but may be higher in urban areas. The amount of CO which is absorbed by the body is dependent on minute ventilation, duration of exposure, and concentrations of CO and oxygen in the surrounding environment [5-8]. After cooking with a gas stove, the indoor air concentrations of CO may reach 100 ppm [9]. Cigarette smokers are exposed to an estimated 400 to 500 ppm of CO while active smoking [10]. Automobile exhaust may contain around 10% (100,000 ppm) CO [11]. Exposure to 70 ppm may lead to Carboxyhemoglobin (CO-Hgb) levels of 10% at equilibrium (approximately 4 hours) [2], while exposure to 350 ppm may lead to CO-Hgb levels of 40% at equilibrium [10]. In addition to these, CO poisoning has been reported in campers using coal stoves in outdoor tents especially during winters, boaters, and people using solvents in paint industry, ice skaters and people using propane-powered resurfacing machines [12].
Neurological manifestations can be acute as well as chronic and depend upon the severity and duration of CO exposure.
Mechanism
Carbon monoxide causes hypoxia by forming carboxyhemoglobin and thereby shifting the oxyhemoglobin dissociation curve to the left [4]. Carbon monoxide’s affinity for hemoglobin is known to be more than 200 times that of oxygen [13], resulting in the formation of carboxyhemoglobin with even relatively low amounts of the inhaled carbon monoxide. Carbon monoxide increases the cytosolic heme levels, which leads to oxidative stress [14], and binds to platelet heme protein and cytochrome c oxidase [13], interrupting cellular respiration [15] and producing reactive oxygen species [16], which in turn leads to neuronal necrosis and finally apoptosis [17]. Impaired cellular respiration provokes stress response, including the activation of hypoxia-inducible factor 1a [18], resulting in neurologic and cardiac protection [19] or injury, dependent on the dose of carbon monoxide, by means of gene regulation. Carbon monoxide exposure also causes inflammation through multiple pathways that are independent of the pathways to hypoxia, resulting in neurologic and cardiac injury.
Clinical features
The clinical manifestations of CO poisoning are variable and severity depends on the concentration of CO in the inspired air, duration of exposure and general health of the involved person. The population at increased risk comprises of infants, elderly, and patients with cardiovascular disorders, lung disorders, anemia and increased basal metabolic rate [20]. The features of acute CO poisoning are better known and more easily recognized than those having chronic exposure. Symptomatology and prognostication correlate poorly with the level of carboxyhaemoglobin measured at the time of presentation. Table 1 shows the clinical features manifested with the varying levels of blood carboxyhaemoglobin concentration [21]. During acute exposure, patients may complain of headache, dizziness, nausea, vomiting, emotional liability, confusion, impaired judgment, clumsiness and syncope. Vomiting may be the only presenting symptom in presenting infants and may be misdiagnosed as gastroenteritis. Coma or seizures can occur in patients with prolonged CO exposure. Elderly people, especially those with coronary disease, may have accompanying myocardial ischemia, which may often result in frank myocardial infarction [22]. CO exposure has deleterious effect on pregnant women because of the greater sensitivity of the foetus to these harmful effects of the gas. The excessive leftwards shift of foetal carboxyhaemoglobin causes more severe tissue hypoxia by releasing comparatively less oxygen to the tissues [23]. Prolonged exposures resulting in altered mental status or coma may be accompanied by retinal hemorrhages and lactic acidosis [24]. Myonecrosis can also occur but it rarely leads to renal failure. Cherry-red colored skin which is associated with severe carbon monoxide poisoning, is seen in around 2-3% of symptomatic cases [25]. Severe poisoning often leads to hypotension and sometimes pulmonary oedema with the former is being the most reliable marker of prognosis.
![]()
Blood CO-Hgb conc (%)
Clinical manifestations
15-20
Mild headache, fatigability
20-30
Impaired motor dexterity, blurred vision, irritability
30-40
Severe muscle weakness, vomiting, mental confusion, delirium
40-50
Tachycardia, irritability
50-60
Seizures, respiratory insufficiency
>60
Coma, respiratory failure, death
Table 1: Levels of CO-Hgb with associated clinical manifestations.
Neuropsychiatric issues may develop insidiously over weeks to months after recovery from CO intoxication. These include intellectual deterioration, memory impairments, cerebral, cerebellar and midbrain damage e.g. Parkinsonism and akinetic mutism with changes in personality like increased irritability, verbal aggressiveness, impulsiveness, violence and moodiness [26]. Around two fifths of patients develop memory impairments and around one third suffer late deterioration of personality [27]. Chronic CO poisoning is often misdiagnosed as chronic fatigue syndrome or chronic infection. Carboxyhaemoglobin levels are usually not excessively elevated in these cases.
Diagnosis
History: Physicians should be alert while dealing with such cases and a high index of suspicion should be kept for cases presenting with history of CO poisoning especially during winters. A meticulous history should involve possible ways of exposure to CO. Patients presenting with flu like symptoms should be asked about use of wood, coal or gas based heating appliances at home or work. A similar symptom occurring in the other housemates is helpful in getting to a diagnosis.
Carboxyhaemoglobin levels: Serum CO-Hgb should be obtained from patients suspected to be having CO poisoning. Low levels may Carboxyhaemoglobin levels: Serum CO-Hgb should be obtained from patients suspected to be having CO poisoning. Low levels may
Lab tests: Other tests include complete blood counts, serum electrolytes, cardiac markers, arterial blood gas analysis (which may show metabolic acidosis because of the combination of hypoxia, cellular respiration inhibition and increased metabolic demand) and serum lactate levels which have been used as a marker for severe poisoning.
Other tests: Chest radiograph may show non cardiogenic pulmonary oedema. Other drug levels showing similar symptoms should be assessed. ECG should be looked for arrhythmias or signs of myocardial infarction.
Neuropsychiatric testing: It includes Mini mental state examination, Weschlar adult intelligence scale, Weschlar memory scale or other more specific tests such as Carbon Monoxide Neuropsychological Screening Battery (CONSB) [28]. Improvement on these tests after oxygen therapy is often considered as an evidence of effectiveness of the therapy.
Brain imaging: CT scan brain may initially show signs of cerebral oedema and later show bilateral basal ganglia hypo densities, particularly in globus pallidus and substantia nigra [29]. MRI may show diffuse white matter involvement predominantly in periventricular areas although basal ganglia, hippocamous and thalamus may also be involved. Usually, patients may show symmetrical T2 and FLAIR hyperintensities in the globus pallidus (Figure 1) and which is often seen to resolve with time and after oxygen therapy [30,31] (Figure 2). Single Photon Emission Computed Tomography (SPECT), quantitative MRI and EEG have been used in CO poisoning but more studies are needed to prove their specificities [32].
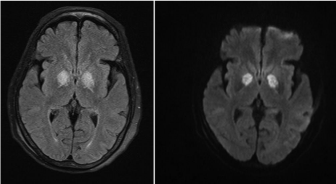
Figure 1: It shows symmetrical T2 and FLAIR hyperintensities in the globus
pallidus.
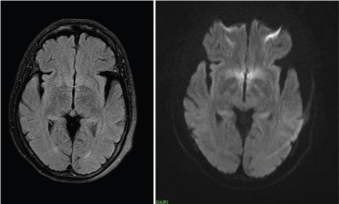
Figure 2: It shows after oxygen therapy.
Management
After the correct diagnosis of CO poisoning, first step should be the maintenance of airway, breathing and circulation. Patient should be advised strict bed rest to reduce oxygen demand and consumption.
Hypoxia is the most common complication of CO poisoning. Oxygen therapy thus is the most important measure to resolve the symptoms. Two types of oxygen therapy using 100% oxygen are used: Hyper Baric Oxygen Therapy (HBOT) and Normo Baric Oxygen Therapy (NBOT). The choice of using the oxygen therapy out of these two is still controversial and lacks a robust data for either of them. In HBOT, oxygen is at a pressure twice to thrice that of atmospheric pressure at sea level whereas it is equal to sea level atmospheric pressure in NBOT. In addition to accelerating the rate of CO elimination from haemoglobin, HBOT also enhances the removal of CO from intracellular binding sites. Its timely administration prevents neuronal injury and prevents delayed neuropsychological sequel by terminating the biological degradation [4]. Despite its benefits, HBOT is associated with adverse effects such as cataracts, reversible myopia, tracheobronchial symptoms, self-limited seizures and barotraumas to the middle ear, the cranial sinuses or the lungs. Another limitation is that not all the hospitals are equipped with such a chamber. For patients with mild CO poisoning (COHb level <20%), a different regime involving 100% NBOT for 6 hours is appropriate.
Other treatment options include targeted temperature management with mild therapeutic hypothermia especially in patients with post cardiac arrest or hypoxic ischaemic brain injury. Administration of sympatholytics may be useful for inhibition of the postganglionic functions of the sympathetic nervous system, thus minimizing the systemic response to acute stressor (CO). Other agents including anti oxidants, steroids, acetyl cholinesterase inhibitors, and erythropoietin have been used but lack a robust data to prove their efficacy in this condition [33].
Patients with CO poisoning should be followed up periodically after discharge. The rate and extent of recovery after poisoning are variable, and recovery is complicated by the development of sequel, which can persist after exposure or develop weeks after poisoning [4] and which can be permanent. Specific therapy for sequel is not available. Such patients should have their symptoms treated, through psychiatric, vocational, cognitive, speech, occupational, and physical rehabilitation, although data on the effects of these interventions in patients with CO related sequel are lacking.
Prevention
Proper public education on the safe operation of heaters, appliances, fireplaces and internal combustion engines is necessary. Burn victims, with an evidence of smoke inhalation from enclosed fire, should undergo testing for COHb levels. During winters, CO poisoning should be suspected in patients presenting with flulike symptoms (e.g., headache, nausea, dizziness), which they may not attribute to a faulty furnace or other heating sources. Carbon monoxide detectors with alarms can improve home and workplace safety [21].
Conclusion
Carbon monoxide is one of the most dreaded and ubiquitous poisons with non specific symptoms. Neurological events are seen in acute as well as delayed and chronic settings. Identification of the source of poisoning should be ascertained and all such cases should be treated as an emergency. 100% oxygen therapy forms the mainstay of treatment. All these patients should have a long term follow up with careful neurocognitive assessment to look for the chronic and permanent neurological sequel.
References
- Kao LW, Nanagas KA. Toxicity associated with carbon monoxide. Clin Lab Med. 2006; 26: 99-125.
- Tomaszewski C. Carbon monoxide. In: Goldfrank LR, Flomenbaum NE, Lewin NA, et al, editors. Goldfrank’s toxicologic emergencies. 7th edition. New York: McGraw-Hill. 2002; 1478-1497.
- Cobb N, Etzel RA. Unintentional carbon monoxide-related deaths in the United States, 1979 through 1988. JAMA. 1991; 266: 659-663.
- Ernst A, Zibrak JD. Carbon monoxide poisoning. N Engl J Med. 1998; 339: 1603-1608.
- Ilano AL, Raffin TA. Management of carbon monoxide poisoning. Chest. 1990; 97: 165-169.
- Olson KR. Carbon monoxide poisoning: mechanisms, presentation, and controversies in management. J Emerg Med. 1984; 1: 233-243.
- Forbes WH, Sargent F, Roughton FJW. The rate of carbon monoxide uptake by normal men. Am J Physiol. 1945; 143: 594-608.
- Roughton FJW. The kinetics of the reaction CO + O2Hb <-> O2 + COHb in human blood at body temperature. Am J Physiol. 1945; 143: 609-620.
- Abelsohn A, Sanborn MD, Jessiman BJ, Weir E. Identifying and managing adverse environmental health effects. Carbon monoxide poisoning. CMAJ. 2002; 166: 1685-1690.
- Raub JA, Mathieu-Nolf M, Hampson NB, Thom SR. Carbon monoxide poisoning- a public health perspective. Toxicology. 2000; 145: 1-14.
- Widdop B. Analysis of carbon monoxide. Ann Clin Biochem. 2002; 39: 378- 391.
- Pelham TW, Holt LE, Moss MA. Exposure to carbon monoxide and nitrogen dioxide in enclosed ice arenas. Occup Environ Med. 2002; 59: 224-233.
- Thom SR. Carbon monoxide pathophysiology and treatment. In: Neuman TS, Thom SR, eds. Physiology and medicine of hyperbaric oxygen therapy. Philadelphia: Saunders Elsevier. 2008: 321-347.
- Cronje FJ, Carraway MS, Freiberger JJ, Suliman HB, Piantadosi CA. Carbon monoxide actuates O(2)-limited heme degradation in the rat brain. Free Radic Biol Med. 2004; 37: 1802-1812.
- Alonso JR, Cardellach F, López S, Casademont J, Miró O. Carbon monoxide specifically inhibits cytochrome c oxidase of human mitochondrial respiratory chain. Pharmacol Toxicol. 2003; 93: 142-146.
- Thom SR, Bhopale VM, Han ST, Clark JM, Hardy KR. Intravascular neutrophil activation due to carbon monoxide poisoning. Am J Respir Crit Care Med. 2006; 174: 1239-1248.
- Piantadosi CA, Zhang J, Levin ED, Folz RJ, Schmechel DE. Apoptosis and delayed neuronal damage after carbon monoxide poisoning in the rat. Exp Neurol. 1997; 147: 103-114.
- Chin BY, Jiang G, Wegiel B, Wang HJ, Macdonald T, Zhang XC, et al. Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc Natl Acad Sci USA. 2007; 104: 5109- 5114.
- Mannaioni PF, Vannacci A, Masini E. Carbon monoxide: the bad and the good side of the coin, from neuronal death to anti-inflammatory activity. Inflamm Res. 2006; 55: 261-273.
- Stewart RD. The effect of carbon monoxide on humans. Annual Review of Pharmacology. 1975; 15: 409-422.
- Mehta SR, Das S, Singh SK. Carbon Monoxide Poisoning. MJAFI. 2007; 63: 362-365.
- National Center for Health Statistics. Vital statistics of the United States, 1988. Washington, DC: Government Printing Office. 1991: 89-1102.
- Farrow JR, Davis GJ, Roy TM, McCloud LC, Nichols GR. Fetal death due to nonlethal maternal carbonmonoxide poisoning. J Forensic Sci. 1990; 35: 1448-1452.
- Ely EW, Moorehead B, Haponik EF. Warehouse workers’ headache: emergency evaluation and management of 30 patients with carbon monoxide poisoning. Am J Med. 1995; 98: 145-155.
- Longo LD, Hill EP. Carbon monoxide uptake and elimination in fetal and maternal sheep. Am J Phsiol. 1977; 232: 324-330.
- Gilbert GJ, Glaser GH. Neurologic manifestations of chronic carbon monoxide poisoning. N Eng J Med. 1959; 264: 1217-1220.
- Smith JS, Brandon S. Morbidity from acute carbon monoxide poisoning at three-year follow-up. Br Med J. 1973; 1: 318-321.
- Messier LD, Myers RA. A neuropsychological screening battery for emergency assessment of carbon-monoxide-poisoned patients. J Clin Psychol. 1991; 47: 675-684.
- Silver DA, Cross M, Fox B, Paxton RM. Computed tomography of the brain in acute carbon monoxide poisoning. Clin Radiol. 1996; 51: 480-483.
- Dubey A, Chouksey D. Carbon monoxide toxicity: A reversible damage to brain. Neurol India. 2017; 65: 672-673.
- Kinoshita T, Sugihara S, Matsusue E, Fujii S, Ametani M, Ogawa T. Pallidoreticular damage in acute carbon monoxide poisoning: Diffusionweighted MR imaging findings. AJNR Am J Neuroradiol. 2005; 26: 1845- 1848.
- Gale SD, Hopkins RO, Weaver LK, Bigler ED, Booth EJ, Blatter DD. MRI, quantitative MRI, SPECT, and neuropsychological findings following carbon monoxide poisoning. Brain Inj. 1999; 13: 229-243.
- Oh S, Choi SC. Acute carbon monoxide poisoning and delayed neurological sequelae: a potential neuroprotection bundle therapy. Neural Regen Res. 2015; 10: 36-38.