
Review Article
Austin Med Sci. 2021; 6(1): 1045.
Cytokines and miRNAs in the Pathogenesis of Rheumatoid Arthritis: A Review
Yu Z1#, Hu Y1#, Liu H2, Fan J1 and Liu W1*
¹Department of Orthopaedics, Affiliated Hospital 2 of Nantong University, Nantong, Jiangsu, China
²School of Clinical Medicine, Nanjing Medical University, Nanjing, Jiangsu, China
#Yu Z and Hu Y contributed equally to this work
*Corresponding author: Wei Liu, Department of Orthopaedics, Affiliated Hospital 2 of Nantong University, 6 Haierxiang Road, Nantong 226001, Jiangsu, China
Received: April 19, 2021; Accepted: May 08, 2021; Published: May 15, 2021
Abstract
Rheumatoid Arthritis (RA) is a chronic autoimmune disease characterized by inflammatory synovial hyperplasia. The pathogenesis of RA may be related to heredity, infection and sex hormones. The initial stage of RA involves the activation of T cells. Immature CD4+ T cells differentiate into T helper (Th) cells and T regulatory (Treg) cells under antigen stimulation and cytokine signal transduction. Cytokines secreted by Th cells and Treg cells play crucial roles in the pathophysiology of RA. The cytokines can be roughly divided into proinflammatory cytokines, anti-inflammatory cytokines, and both pro- and antiinflammatory cytokines. The imbalance between pro-inflammatory cytokines and anti-inflammatory cytokines would lead to a variety of autoimmune diseases. The disease severity was significantly indicated by serum or plasma cytokine levels with RA patients. Many clinical trials have shown that anticytokine drugs are effective in treating RA. This article reviews the differentiation process of different Th cells and Treg cells, the roles of cytokines secreted by them in the pathogenesis of RA and how miRNAs mediate immune regulation in RA. By understanding the roles of cytokines and miRNAs in the pathogenesis of autoimmunity, it is necessary to develop potential anti-cytokine drugs and biomarkers/therapeutic targeted drugs through various ways in the treatment of RA.
Keywords: Rheumatoid arthritis; T helper cells; T regulatory cells; Proinflammatory cytokines; Anti-inflammatory cytokines; miRNA
Abbreviations
RA: Rheumatoid Arthritis; Th cells: T helper cells; Treg cells: T regulatory cells; Foxp3: Forkhead Box Protein 3; IFN: Interferon; IL: Interleukin; T cells: T lymphocytes; APC: Antigen-Presenting Cells; MHC-II: Major Histocompatibility Complex-II; STAT: Signal Transducer and Activator of Transcription; IL-12R: IL-12 Receptor; CXCR: CXC Motif, Receptor; CCR: C-C Chemokine Receptor; IP10: IFN-γ-Inducible 10-kDa Protein; Mig: Monokine Induced by IFN-γ; I-TAC: IFN-Inducible T cell Alpha Chemoattractant; Smads: Smad Family Proteins; TGF-ß: Transforming Growth Factor-ß; RORγt: Retinoid-Related Orphan Receptor γt; IRF4: Interferon Regulatory Factor 4; nTregs: natural Tregs; iTregs: induced Tregs; TIGIT: T cell Ig and ITIM domain; CIA: Collagen-Induced Arthritis; G: Guanine; A: Adenine; MSCs: Mesenchymal Stem Cells; IL-6Ra: IL6-specific Receptor a; sIL-6R: Soluble IL-6R; CSIF: Cytokine Synthesis Inhibitory Factor; vIL-10: Viral IL-10; EBI3: Epstein-Barr virus-Induced Gene 3; EAE: Experimental Autoimmune Encephalomyelitis; PIAS3: Protein Inhibitor of STAT3; VEGF: Vascular Endothelial Growth Factor
Introduction
Rheumatoid Arthritis (RA) is a chronic autoimmune disorder characterized by nonspecific inflammation of synovial membranes and joints. Accompanied by extraarticular organ involvement and serum rheumatoid factor positive, RA would lead to joint deformity and loss of function. Increasing with considerable morbidity and mortality worldwide, RA is affecting 0.5-1.0% of the general population [1]. A heavy economic pressure was brought and a social burden on the whole world by RA.
As a critical role in host defense, CD4+ T cells make major contributions to the generations of autoimmune and inflammatory diseases. Naive CD4+ T cells were differentiated into various forms of T helper (Th) cells and T regulatory (Treg) cells under such various conditions as antigenic stimulation and cytokine signaling. Since the discovery of the Th1/Th2 dichotomy by Robert Coffman and Timothy Mossman in 1986, many Th subsets were discovered. Each Th subset was found with a unique cytokine profile, functional properties and distinctively presumed roles in the autoimmune tissue pathology. Treg cells play an important role in regulating the immune response. Treg cells were lately discovered in regulating Forkhead box protein 3+ (Foxp3+) as key molecules. Another significant member of the CD4+ T cell subsets is Th17 cell; it plays a critical role in autoimmune diseases. In recent years, with the help of modern technology, the knowledge of Th17 cells was with tremendous development. Th17 cells also contribute to the pathogenesis of arthritis by modulating antibody function [2]. Manipulation of dysregulated miRNAs in vivo through miRNA delivery or inhibition offered a promise for new therapeutic strategies in treating rheumatic diseases [3].
CD4+ T cells & Th cells
In 1986, Robert Coffman and Timothy Mossman had a revolutionary discussion over two types of Th cells, Th1 and Th2 cells, based on the cytokine secreted by distinct CD4+ T cell subsets [4], Interferon-γ (IFN-γ) and interleukin-4 (IL-4), respectively [5]. In recent years, the classical Th1/Th2 models were hugely modified and the mechanisms of Th cell differentiation were deeply uncovered.
Originated from lymphoid cells of marrow, T lymphocytes (T cells) maturate in thymus under complicated and rigorous regulations. Exiting from thymus, matured T cells carry all the genetic components to recognize antigens and express CD4 or CD8 molecules. Exogenous antigens are ingested by Antigen-Presenting Cells (APC) through phagocytosis or pinocytosis to form phagosomes. Phagosome was fused with lysosomes to form phagolysosomes. Antigens are degraded into small molecular polypeptides by proteolytic enzymes in acidic environment of phagolysosomes, among which antigen peptides are immunogenic. After entering the Golgi apparatus, the Major Histocompatibility Complex-II (MHC-II) molecules synthesized in the endoplasmic reticulum are carried by secretory vesicles and fuse with phagolysosomes to form an antigen-peptide-MHC-II complex. The complex expressed on the surface of APC can be identified and bound by corresponding CD4+ T cells. Through a complex process of blast cell transformation, the activated T cells can undergo further differentiation to various subtypes of T cells and can express different combinations of immune response genes [6].
Th1 cells
Th1 cell differentiation is generally induced by IL-12 secreted from APCs. However, IFN-γ mRNA, and only modestly augments antigen-induced IFN-γ mRNA cannot be induced by IL-12 itself from Th1 cells [7]. IFN-γ and IL-12 cytokines activate signal transducer and activator of transcription 1 (STAT1) and STAT4 respectively to promote the expression of T-bet, IFN-γ and IL-12 receptor (IL-12R) ß2 [8-10]. T-bet, discovered as a novel protein belonging to the T box family by Szabo et al. in 2000 [11], is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells [9]. T-bet was served as a master regulator for Th1 cell differentiation [11]. The expression of T-bet resulted in Th1 cell development and the specific expression of IFN-γ [11] as well as IL-12Rß2. The production of IFN-γ and IFN-γ-STAT1-T-bet pathway construct a positive feedback loop. IFN-γ- STAT1-T-bet pathway was considered as the main mechanism of Th1 cell differentiation. It is worth mentioning that Lighvani et al. firstly found that the IFN-γ-STAT1-T-bet pathway serves as a powerful amplification mechanism in Th1 cell differentiation in vitro [12]. However, the IL-12-STAT4-T-bet pathway is critical both in vitro and in vivo to Th1 cell differentiation [13]. The differentiation and secretion processes of Th cells and Treg cells were illustrated in Figure 1.
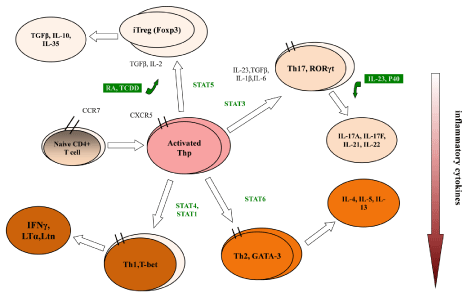
Figure 1: The differentiation and secretion of Th cells and Treg cells. Naive CD4+ T cells differentiate into Th1, Th2, Th17 and Treg cells under antigen stimulation
and cytokine signal transduction. Cytokines secreted by different Th cell subgroups and Treg cells are distinctive.
In addition to IL-12 and IFN-γ mentioned above, IL-2 and TNFa are two well-known cytokines secreted by Th1 cells. Another two specific kinds of cytokine receptors expressed by Th1 cells are CXCR3 (CXC motif, receptor 3) and CCR5 (C-C chemokine receptor 5) [14,15]. CXCR3 is the receptor for the CXC chemokines IFN-γ- inducible 10-kDa protein (IP10), Monokine induced by IFN-γ (Mig) and IFN-inducible T cell Alpha Chemoattractant (I-TAC). CCR5 was first cloned from a human genomic DNA library based on its similarity to a murine C-C chemokine receptor clone in 1996. CXCR3 is present in most peripheral blood memory T cells and is expressed at higher levels in Th1 cells than in Th2 cells [16,17].
Th2 cells
The differentiation of Th2 cells abides by a similar process to Th1 cell development. IL-4 was first identified as an essential cytokine for Th2 cell differentiation. IL-4 and IL-2 cytokines activate STAT6 and STAT5 respectively to promote the expression of transcription factor GATA-3 and cMaf [18,19]. The expression of cMaf can enhance the production of IL-4, IL2Ra and IL4Ra. GATA3 serves as the first master regulator for Th2 cell differentiation [20]. In a word, STAT6 and STAT5 were activated by IL-4 and IL-2 cytokines respectively. The expression of IL-4 and IL-4-STAT6-GATA3 pathway also constructs a positive feedback loop. The feedback loop was considered as the main mechanism of Th2 cell differentiation.
In contrast to the secretion of CXCR3 and CCR5 by Th1 cells, Th2 cells were found to possess a specific chemokine receptor CCR4. CCR4 is a small, basic, structure-related molecule that regulates cell transport of various types of leukocytes by interacting with 7-transmembrane G protein-coupled receptors [21]. CCR4 was found to be strongly expressed in human T cell lines and peripheral blood T cells, whereas not in B cells, natural killer cells, monocytes nor granulocytes [22].
Th17 cells
In 2005, Laurie Harrington firstly found that Th17 cells were distinct from the T helper types 1 and 2 lineages [23]. The differentiation mechanism of Th17 cells went through a tortuous process. Initially, as a crucial cytokine, IL-23 triggers the STAT3 pathway to promote the secretion of IL-17A via its distinct IL-23R [23-25]. Subsequently, Th17 cells were highly generated when with Smad family Proteins (Smads) and STAT3, activated by transforming growth factor-ß (TGF-ß) and IL-6 respectively [26-28]. Then, the expression of transcription factor retinoid-related orphan receptor γt (RORγt) was induced to initialize Th17 cell differentiation [29].
However, until 2010, Ghoreschi et al. showed that Th17 cell differentiation could proceed in the absence of TGF-ß signaling [30]. Another important cytokine involved in the differentiation of Th17 cells is IL-1ß. IL-1ß signaling promoted Th17 cell function by induction to transcription factor RORγt and Interferon Regulatory Factor 4 (IRF4) [31]. A recent study found that IL-21 could substitute for IL-6 to generate Th17 cells in vitro [32]. Th17 cells then produce effector cytokines IL-17A, IL-17F, IL-21, IL-22 and TNFa, among which IL-17A and IL-17F are mainly produced. Effector T cells in patients with RA are hyporesponsive due to chronic exposure to TNFa [33]. IL-21 further promotes the process of Th17 cell differentiation in a positive feedback manner, which may be similar to the roles of IFN-γ and IL-4 in the development of Th1 and Th2 cells, respectively.
Treg cells
CD4+ Tregs are composed of two types: natural Tregs (nTregs), which originate from the thymus and induced Tregs (iTregs), which are generated in the periphery. The nTregs play a role through contacts between non-cytokine dependent cells, but the iTregs work by employing the expression of inhibitory cytokines such as IL10, IL- 35 and TGF-ß. Differentiation environment, antigen specificities and inhibition mechanism determine the direction of differentiation of Treg cells.
In 1995, the CD25 molecule (the IL-2 receptor a-chains) was identified as the first surface marker for Tregs by Sakaguchi and his colleagues [34]. Subsequently, as a forkhead transcription factor encoded by the X chromosome, Foxp3 was indicated as a sign of Treg cells, critical in both thymic Treg cell differentiation and peripheral Treg cell differentiation [35,36]. More recently, CD127 was regarded as a candidate for Treg cells. In 2007, it was found that STAT5 activated by IL-2 is required for the development of Foxp3+ Treg cells, STAT5 binds directly to the transcription factor Foxp3 to induce the Treg cell differentiation [37]. However, it was found that the differentiation of iTregs is induced not only by stimulation with IL-2 but also by activation of TGF-ß [38]. Immature CD4 single-positive thymocytes receive TCR signals of varied strength via interactions with peptide-MHC on antigen-presenting cells [38]. Beyond TCR, such co-stimulatory factors as CD28, CD80/86 (B7), CD40 and IL2Rß are also essential for nTreg cell differentiation [39-41].
As mentioned above, iTreg cells are generated in the periphery. However, TCR repertoires displayed that most of the peripheral Treg cells originate in the thymus [42]. In 2007, it was indicated that both thymic and peripheral Foxp3- non-Treg cells expressed TCRs distinct from Foxp3+ Treg cells through experiments conducting in BDC2.5 TCR transgenic mice [43]. Results indicated that most Treg cells in the periphery are limited to particular environments or tissues. An imbalance between Foxp3 Treg cells and Th17 cells is often linked with autoimmune diseases, including arthritis [2].
Foxp3 is not sufficient for the acquisition of a stable Treg phenotype since human naive T-cells can readily express Foxp3 upon TCR stimulation [44]. The sustained expression of high levels of Foxp3 in iTreg cells is influenced by a particular mode of TCR signaling and through synergistic effect with such signals as TGF-ß and IL-2. TGF- ßR signaling appears to be required for most induction to Foxp3 in peripheral CD4+ T cells [45]. IL-2 is also required for TGF-ß-mediated induction to Foxp3 in peripheral T cells in vitro [46]. IL-2 not only activates STAT5 for the development of Foxp3+ Treg cells but also opposes CD4+ T cells differentiated into Th17 cells [47].
Another important factor that has been demonstrated to exert direct effects on iTreg cell differentiation is the retinoic acid. It has been documented that the all-trans retinoic acid can assist TGF-ß in converting naive T-cells into Foxp3+ cells with potent suppressive activity by CD3/CD28 stimulation [48].
Different types and strength of TCR stimulation determine that CD4+CD25+ Treg cells express whether high levels of bound or soluble TGF-ß. Several cell-surface molecules were proposed to play a role as mediators of Treg cell-mediated suppression, such as CD25, CD39, CD73, CD152 (CTLA-4) and a recent discovery of a novel Ig family member, T cell Ig and ITIM domain (TIGIT) [49-52]. When Treg cells interact with dendritic cells, TIGIT seems to induce dendritic cells to produce immunosuppressive cytokines IL-10 and TGF-ß [53]. It was found that although retinoic acid augments Foxp3 induction inhibited the expression of IL-10. The fact indicated that one potential existing distinct differentiation cues for iTreg cells and IL-10-producing Treg cells [54]. However, it is yet unclear what the relative contributions of iTreg cells are to the mature peripheral Treg cell population, further studies will be important for dissecting the functional niches of these cells. Admittedly, the imbalance between Foxp3+ Treg and Th17 cells is often linked with RA [2].
It has now been shown that several of the drugs employed in the medical therapy of RA can partially restore Treg cell function, which has also been associated with amelioration of the clinical symptoms of RA [55]. CRISPR-mediated Treg genome editing was considered as a potential application of personalized therapy of RA [56].
Roles of Cytokines in RA
Secreted by Th cells and Treg cells, cytokines play crucial roles in such biological processes as cell growth, proliferation, differentiation, tissue repair, inflammation and regulation of the immune response [57]. The cytokines secreted by Th1, Th2, Th17 and Treg cells can be classified three types: pro-inflammatory cytokines, anti-inflammatory cytokines and both pro- and anti-inflammatory cytokines. TNFa, IL- 17 and IL-21 are pro-inflammatory cytokines. IL-4, IL-10, IL-13 and IL-35 are anti-inflammatory cytokines. IL-6 is a kind of both proand anti-inflammatory cytokines. IL-38 was reported to exert an anti-inflammatory function. The balance between pro-inflammatory cytokines and anti-inflammatory cytokines was illustrated in Figure 2.
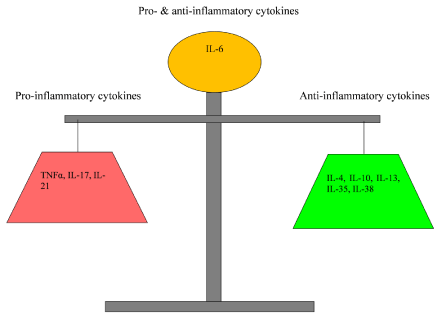
Figure 2: The balance between pro-inflammatory and anti-inflammatory cytokines. Pro-inflammatory cytokines and anti-inflammatory cytokines maintain a dynamic
balance in the human internal environment. The up-regulation of pro-inflammatory cytokines and down-regulation of anti-inflammatory cytokines would lead to the
occurrence of RA.
TNFa
TNFa is an important pro-inflammatory cytokine with diverse functions in the immune system [58]. It was confirmed that anti- TNFa had therapeutical effects on joint pathology in both Collagen- Induced Arthritis (CIA) models of DBA/1 mice and RA patients [59,60]. Although TNFa was insufficient for the initiation step of Th17 cell differentiation, it was found to amplify the differentiation process in vitro [61]. On one hand, TNFa promotes the process of Th17 cell differentiation induced by IL-6 and TGF-ß; on the other hand, TNFa indirectly promotes the development of Th17 by inducing IL-6 [61]. Interestingly, the IL-17 cytokine is a strong inducer of TNFa, as if they together form a positive feedback loop. In the last decade, many scholars studied the relationship between TNFa and RA from the perspective of molecular biochemistry. The transition of Guanine (G) to Adenine (A) at position 308 is the first identified polymorphism. Several studies have reported the association between the -308 TNF SNP and the susceptibility to RA [62]. The enhancements of TNFa production and thus the disease susceptibility were also associated with gene polymorphisms at positions -857 C/T, -863 C/A, -1031 T/C and +1304 G/A [63].
IL-4
IL-4 is an essential cytokine for Th2 cell differentiation, expression and secretion. It activates STAT6 to promote the expression of transcription factor GATA-3, the full development of Th2 cells and then the specific expression of IL4. In experimental animal models, it was found that IL-4 inhibits the production of inflammatory cytokines IL-1 and IL-6 in joints, thereby inhibiting the production of arthritis [64]. In a synovial fluid of some RA patients, serving as an anti-inflammatory cytokine, IL-4 hinders the formation of proinflammatory cytokines such as rheumatoid factor TNFa, IL-1ß and IL-6 [65]. “Apoptosis resistance” was believed as the leading cause of aberrant cell survival in RA [55]. Studies in vitro showed that IL-4 reduces bone resorption by directly affecting osteoclasts and inhibiting the production of MMPs [65]. As the cross-regulation between Th17 and Th2 cells, IL-4 can inhibit Th17 cell differentiation and IL-17 induction [23]. In contrast, when IL-4 was blocked and independent of Th1 or Th2-related transcription factors (T-bet, STAT-1, STAT-4 and STAT-6), production capacity of Th17 cells increase in vitro [66]. One meta-analysis summarized that promoter polymorphism (590, T/C) of IL-4 was a useful marker for early diagnosis and treatment of RA [67].
It was reported that the potential role of vitamin D were provided a fresh perspective on RA pathogenesis and treatment [68]. To alter the frequency and function of memory lymphocytes while promoting Treg cell generation, Mesenchymal Stem Cells (MSCs) have been proposed as a candidate of choices for RA cell therapy [69].
IL-6
IL-6 cytokine is produced in many immune and stromal cells and has both pro- and anti-inflammatory properties in the immune system [70]. In the classic signaling pathway, IL-6R has two chains, namely IL6-specific receptor a (IL-6Ra) and a signal transducer (gp130). IL-6 binds to transmembrane IL-6Ra, which then associates with gp130 to activate STAT1 and STAT3 for downstream biological activities [71]. However, in IL-6 trans-signaling, soluble IL-6R (sIL-6R) lacking transmembrane and cytoplasmic regions binds to gp130 subunits. Evidence suggests that classic signaling is anti-inflammatory whereas trans-signaling is pro-inflammatory [72]. The pleiotropic cytokine IL-6 is crucial to the differentiation of Th17 cells, and the balance between pathogenic Th17 and protective Treg and that targeting the IL-6R by humanized antibodies provided new insights to improve RA [73].
IL-10
IL-10, known as human Cytokine Synthesis Inhibitory Factor (CSIF), is a kind of anti-inflammatory cytokine. It is produced by such innate and adaptive immunity cells as dendritic cells, macrophages, mast cells, natural killer cells, eosinophils, neutrophils, B cells, CD8+ T cells, Th1 cells, Th2 cells, Th17 cells and Treg cells [74]. However, IL-10 is mainly secreted by Th2 and Treg cells. It was found that Th17 cells driven by TGF-ß and IL-6, were shown to express IL-10 [75]. On the one hand, IL-10 reduces such pro-inflammatory cytokines as TNFa, IL-1a, IL-1ß, IL-6, IL-8, IL-12 and GM-CSF [65]. Dendritic cells promote the secretion of IL-4 and reduce the production of IL- 12, resulting in Th1/Th2 imbalance in an IL-10-dependent manner [76]. IL-10 inhibits Th17 by reducing STAT3 phosphorylation and RORγt [77].
Mouse DBA1 CIA models showed that the adenovirus-mediated gene transfer and the expression of viral IL-10 (vIL-10) significantly reduced the severity of arthritic symptoms; while the pretreatment of Ab anti-IL-10 can abolish the effect of vIL-10 gene transfer on the development of CIA [78].
IL-10 deregulation plays a role in the development of such inflammatory diseases as neuropathic pain, Parkinson’s disease, Alzheimer’s disease, osteoarthritis, rheumatoid arthritis, psoriasis, systemic lupus erythematosus, type 1 diabetes, inflammatory bowel disease and allergy [74].
IL-13
IL-13 is a kind of anti-inflammatory cytokine mainly produced by Th2 cells. IL-13 shares a common cellular receptor (IL-4 type 1 receptor) with IL-4 [79]. IL-13 decreases the production of pro-inflammatory cytokine TNFa and IL-6 (both pro- and antiinflammatory cytokine). As a cytokine secreted by Th2 cells like IL-4 and IL-10, IL-13 has been found similar effects on down regulating the production of IL-1ß [65]. The ability of IL-13 to down-regulate cytokine secretion is equivalent to that of IL-4, but not as good as that of IL-10 [65]. However, adding IL-10 to the cultures treated with IL-4 or IL-13 reduces the production of IL-1ß [65].
The IL-13 cytokine expresses higher than IL-4 in SF indicated that IL-13 plays a more important role in the regulation of synovial inflammatory responses in patients of RA [80]. Two groups of experiments conducted on CIA mice suggested that IL-13 inhibits bone resorption by suppressing cyclooxygenase-2-dependent prostaglandin synthesis in osteoblasts and subcutaneous inoculation of IL-13-secreting vector cells can significantly alleviate collageninduced arthritis [81,82].
IL-17
IL-17 is a kind of pro-inflammatory cytokine mainly secreted by Th17 cells. IL-17 family contains six cytokines: IL-17A, IL-17B, IL- 17C, IL-17D, IL-17E and IL-17F. Characteristic cytokines of Th17 cells, IL-17RA and IL-17RC are the corresponding receptors of IL- 17A and IL-17F. After binding, the heterodimeric receptor complex initiates intracellular signaling pathways. As IL-17F shares about 50% homology in amino acid sequence with IL-17A [83], they are similarly associated with autoimmune disorders. The differentiation of Th17 cells is controlled by some cytokines (IFN-γ, IL-1ß, IL-4, IL-6, IL-21 and TGF-ß) and transcription factors (STAT1, STAT3, STAT4, STAT6, T-bet, GATA-3, RORγt and RORa) [84].
The IL-17 level of RA patients is higher than that of healthy volunteers and osteoarthritis patients [85,86]. IL-17 increases the production of some pro-inflammatory cytokines such as TNFa, IL1ß and IL-6 [87]. A group of experiments conducted in DBA- 1 mice indicated that the inflammation of synovial membrane and the damages to joint aggravated when IL-17 was over-expressed in the keen joint of collagen type II immunized mice [85,88]. However, the joint inflammation, cartilage destruction and bone erosion are ameliorated when IL-17A is neutralized by anti-murine IL-17 antibody [89]. Moreover, IL17A-deficient mice loss of the ability to activate T cells and are protected from the CIA [90]. However, compared to IL- 17A, IL-17F contributes less to the arthritis pathogenesis of RA.
IL-21
IL-21 is a kind of pro-inflammatory cytokine mainly produced by Th17 cells and Th cells [32]. Belonging to the common γ-chain family, IL-21 is homologous to IL-15, IL-2 and IL-4 [91]. According to the process of Th17 differentiation described above, either IL-6 plus TGF-ß or IL-21 plus TGF-ß would activate some transcription factors to initialize Th17 cell differentiation. It was found that IL-21RFC chimeric protein significantly reduces the severity of the RA disease in CIA mice and AA rats [92]. However, two groups of experiments conducted in Experimental Autoimmune Encephalomyelitis (EAE) models demonstrated that the differentiation of Th17 cells was greatly affected in the absence of IL-6 rather than IL-21 [93]. It was suggested that IL-21 plays a supporting role in the process of Th17 cell differentiation, but not indispensable.
IL-35
As a heterodimer of IL-12 family, IL-35 is found as antiinflammatory cytokine secreted by CD4+ Foxp3+ Treg cells. IL-35 consists of the IL-12a (P35) and the Epstein-Barr virus-induced gene 3 (known as EBI3) subunits [94]. P35 and EBI3, which are binding to the IL-35 receptor IL-12Rß2 and gp130 respectively, would lead to the activation of STAT1 and STAT4 proteins [95]. It was shown that T cells that expressed IL-12Rß2 induced only phosphorylated STAT4 in response to IL-35, whereas T cells that expressed gp130 induced only phosphorylated STAT1 [95]. The regulatory activity of Treg cells decreased in vitro and in vivo of EBI3 or P35 deficient mice, which indicated that the expression of IL-35 is necessary to achieve the optimal function of Treg cells [95].
It was demonstrated that the treatment with IL-35 could ameliorate the severity of CIA mice although further studies are required to clarify its roles in RA [96,97]. However, the mechanisms of how IL-35 alleviates arthritis are different in the two groups. In one CIA model, it was found that IL-35 inhibited the production of IL- 17 and enhanced the synthesis of IFN-γ [97]. In another CIA model, IL-35 treatment increased the number of Treg cells and IL-10, and it suppressed the development of Th1 and Th17 cells [96].
IL-35 induces the expansion of Tregs and mediates the suppression of Th17 cell differentiation. IL-35 also upregulates osteoprotegerin and suppresses RANKL, thus inhibiting bone resorption. Thus, IL- 35 was thought of as a potential immunomodulator in autoimmune rheumatic diseases [98].
IL-38
As a newly discovered IL-1 family cytokine, IL-38 is expressed in several tissues and secreted by various kinds of cells. IL-38 was reported to exert an anti-inflammatory function. The IL-38 family is a composite of IL-36R, IL-1RAPL1 and IL-1R1 to block binding with other pro-inflammatory cytokines and to inhibit subsequent signaling pathways [99]. Inflammatory autoimmune diseases are characterized by an imbalance between Ths, especially Th1s, Th17s and Tregs [100]. Many abnormal expressions of IL-38 in inflammatory autoimmune diseases (rheumatoid arthritis, inflammatory bowel disease, systemic lupus erythematosus, psoriasis) were found to be associated with Th1s, Th17s and Tregs [101]. IL-38/IL-36R and/or IL-38/IL-1RAPL1 axis primarily play an anti-inflammatory role in the development and resolution of inflammatory autoimmune diseases, and IL-38/IL-36R and/or IL-38/IL-1RAPL1 axis was indicated a possible therapeutic benefit of IL-38 in these diseases [101].
miRNAs Mediate Immune Regulation in RA
miRNAs are a class of small noncoding RNAs that are only 21-25 nucleotide long. Many studies have shown that miRNA dysfunction is closely related to RA. The initiation of RA pathogenesis is activation of the innate immunity mediated by DCs following recognition of exogenous stimuli or autoantigens. Aberrations in miR-34a in DCs may predispose to RA development. A recent study has shown that CD1c+ DCs from RA patients are hyperactivated with elevated miR-34a expression in association with a reduced level of the tyrosine kinase receptor AXL, an important inhibitory DC autoregulator and a direct target of miR-34a [102]. Over-expression and gene silencing of miR-34a in human DCs confer respective hyperand hypo-production of inflammatory cytokines including TNF and IL-6 upon TLR stimulation. Moreover, miR-34a deficient mice are resistant to collagen-induced arthritis with reduced DC-T cell interaction in vivo. Interestingly, there is a selective impairment in the induction to Th17 in miR-34a-/- mice, suggesting that miR-34a is critical to arthritis induction, at least in part, via DC-induced Th17 expansion. It was showed that miR-34a is an epigenetic regulator of DC function that may contribute to RA [102].
Indeed, the higher Th17 frequency and/or the higher Th17/Treg ratio have been observed in RA patients [103,104]. On the other hand, the down-regulation of miR-21 in RA patients would lead to an increase in the Th17/Treg ratio [105]. miR-21 was verified as MaR1 downstream miRNA, which was upregulated by MaR1, modulating the Treg/Th17 balance and thus ameliorating the RA progression. Thus, MaR1 was argued as a therapeutic target for RA, likely operating through effects on the imbalanced Treg/Th17 ratio found in the disease [105]. Maresin 1 improves the Treg/Th17 imbalance in RA through miR-21 [105]. The elevated Th17 frequency of RA patients was showed to correlate with an overexpression of miR-301a-3p, whereas an inverse relationship with its potential target protein inhibitor of STAT3 (PIAS3) [106]. Dendritic cells from RA patients’ peripheral blood induce Th17 cell differentiation via miR- 363/Integrin av/TGF-ß axis [104].
The elevation of miR-16 was correlated with Th17/Treg cell imbalance and high Th17 cell frequency in RA patients [107]. Overexpression of miR-498 suppresses Th17 cell differentiation in RA patients [108]. miR-16 participates in the occurrence and development of RA by regulating the expression of such cytokines as TNFa, IL-8, IL-6 and IL-4, and affects the proliferation and differentiation of Th17 cells and Treg cells as well [109]. Upregulated expression of miRNA-16 correlates with Th17/Treg cell imbalance in patients with RA [107]. miR-301a-3p and Th17 cells play an important role in the process of RA [106]. Decreased expression of miRNA-21 correlates with the imbalance of Th17 cells and Treg cells in patients with RA [103].
miRNA-146a-/-Foxp3+ Treg cells mediate IFN-γ-dependent lesions in a variety of organs because of STAT1 expression [110]. These Tregs produce such inflammatory cytokines as IFN-γ, TNFa and IL-17 [111]. The diminished expression of miRNA-146a upon stimulation in Treg cells of RA patients correlates inversely with disease activity and its direct target, STAT1 expression [112]. Treg/ inflammatory T cell plasticity is promoted by the inflammatory milieu in autoimmune conditions as well as miRNA-146a insufficiency [113]. miRNA-155 modulation was thought of as a potential therapy for RA [114]. miRNA-155 played an essential role in the pathogenesis of autoimmune arthritis in mice [115]. Decreased expression of miRNA-146a and miRNA-155 contributes to an abnormal Tregs phenotype in patients with RA [112]. In all, miRNA-155 is a sort of important miRNAs in RA pathogenesis.
That transfection of miRNA-155 inhibitors to B cells from RA patients increased supported the functional interference of miRNA-155 in RA. Similarly, miRNA-155 is also upregulated in RA synovial monocytes and macrophages [116]. miRNA-155 was showed highly expressed in B cells from synovial tissues of RA patients [117]. Overexpression of miRNA-155 in RA monocytes also leads to higher expression of such chemokines as CCL3, CCL4, CCL5 and CCL8 [118]. Overexpression of miRNA-155 in monocytes leads to a downregulation of the negative regulator SHIP-1 and an increase in TNFa, IL-6 and IL-1ß [119]. miRNA-10a down-regulation also accelerates the NF-κB-dependent pathway involving IκB, IRAK4 and TAK1. The up-regulation of miRNA-143 and miRNA-145 in RA synoviocytes renders the cells more sensitive to TNFa and Vascular Endothelial Growth Factor (VEGF) stimulation [120].
Conclusion
Although the exact pathogenesis of RA is not yet clear, the increase of pro-inflammatory cytokines and the decrease of anti-inflammatory cytokines play an important role in the pathophysiology of RA. Great efforts have been made to study the pathogenesis of RA, but the treatment was still limited to alleviating the inflammatory reaction of joints, delaying irreversible bone destruction, protecting the functions of joints as much as possible and ultimately achieving the goal of reducing disease activity. Biological agents that have been used in the clinic include Infliximab, Etanercept, Adalimumab, Tocilizumab and Rituximab, which make the condition of most patients with RA well controlled or even completely alleviated. Because cytokines are key factors in RA, it is necessary to develop more cytokine-targeted therapies and ensure that these drugs are both effective and safe in the treatment of patients with RA. Besides, if we use multiple cytokinetargeted therapies, this will undoubtedly be very gratifying. miRNAs act a critical role in autoimmunity, specifically on their regulatory roles in the pathogenesis of several rheumatic diseases including systemic lupus erythematosus, rheumatoid arthritis and spondyloarthritis. Thus, based on cell therapeutic approaches, miRNAs were thought of as potential biomarkers/therapeutic targets for future treating RA.
Highlights
• CD4+ T cells can differentiate into various forms of Th cells and Treg cells.
• Cytokines can be classified as three types according to their inflammation.
• The imbalance between pro-inflammatory and antiinflammatory cytokines leads to RA.
• Many studies show that miRNA dysfunction is closely related to autoimmune diseases.
• miRNAs can be viewed as potential biomarkers/therapeutic targets for treating RA.
Acknowledgment
We thank Papertrans (Beijing) Technology Service Co., Ltd. (www.papertrans.cn) for its linguistic assistance during the preparation of this manuscript. All references were retrieved from PubMed and Google Scholar.
References
- SA Fazal, M Khan, SE Nishi, F Alam, N Zarin, MT Bari, et al. A Clinical Update and Global Economic Burden of Rheumatoid Arthritis. Endocr Metab Immune Disord Drug Targets. 2018; 18: 98-109.
- N Komatsu, H Takayanagi. Immune-bone interplay in the structural damage in rheumatoid arthritis. Clin Exp Immunol. 2018; 194: 1-8.
- IKY Lam, JX Chow, CS Lau, VSF Chan. MicroRNA-mediated immune regulation in rheumatic diseases. Cancer Lett. 2018; 431: 201-212.
- TR Mosmann, H Cherwinski, MW Bond, MA Giedlin, RL Coffman. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986; 136: 2348-2357.
- SL Swain, AD Weinberg, M English, G Huston. IL-4 directs the development of Th2-like helper effectors. J Immunol. 1990; 145: 3796-3806.
- SL Bevington, P Cauchy, DR Withers, PJ Lane, PN Cockerill. T Cell Receptor and Cytokine Signaling Can Function at Different Stages to Establish and Maintain Transcriptional Memory and Enable T Helper Cell Differentiation. Front Immunol. 2017; 8: 204.
- J Yang, TL Murphy, W Ouyang, KM Murphy. Induction of interferon-gamma production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol. 1999; 29: 548-555.
- WE Thierfelder, JM van Deursen, K Yamamoto, RA Tripp, SR Sarawar, RT Carson, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996; 382: 171-174.
- M Afkarian, JR Sedy, J Yang, NG Jacobson, N Cereb, SY Yang, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002; 3: 549-557.
- T Usui, R Nishikomori, A Kitani, W Strober. GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL- 12Rbeta2 chain or T-bet. Immunity. 2003; 18: 415-428.
- SJ Szabo, ST Kim, GL Costa, X Zhang, CG Fathman, LH Glimcher. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000; 100: 655-669.
- AA Lighvani, DM Frucht, D Jankovic, H Yamane, J Aliberti, BD Hissong, et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci USA. 2001; 98: 15137-15142.
- MH Kaplan, YL Sun, T Hoey, MJ Grusby. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996; 382: 174-177.
- M Samson, O Labbe, C Mollereau, G Vassart, M Parmentier. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry. 1996; 35: 3362-3367.
- M Loetscher, P Loetscher, N Brass, E Meese, B Moser. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998; 28: 3696-3705.
- F Sallusto, D Lenig, CR Mackay, A Lanzavecchia. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998; 187: 875-883.
- R Bonecchi, G Bianchi, PP Bordignon, D D’Ambrosio, R Lang, A Borsatti, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998; 187: 129-134.
- W Zheng, RA Flavell. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997; 89: 587-596.
- G Le Gros, SZ Ben-Sasson, R Seder, FD Finkelman, WE Paul. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J Immunol. 2008; 181: 2943-2951.
- DH Zhang, L Cohn, P Ray, K Bottomly, A Ray. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2- specific expression of the interleukin-5 gene. J Biol Chem. 1997; 272: 21597-21603.
- L Rivino, M Messi, D Jarrossay, A Lanzavecchia, F Sallusto, J Geginat. Chemokine receptor expression identifies Pre-T helper (Th)1, Pre-Th2, and nonpolarized cells among human CD4+ central memory T cells. J Exp Med. 2004; 200: 725-735.
- T Imai, M Baba, M Nishimura, M Kakizaki, S Takagi, O Yoshie. The T celldirected CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J Biol Chem. 1997; 272: 15036-15042.
- LE Harrington, RD Hatton, PR Mangan, H Turner, TL Murphy, KM Murphy, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005; 6: 1123- 1132.
- H Park, Z Li, XO Yang, SH Chang, R Nurieva, YH Wang, et al. A distinct lineage of CD4+ T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005; 6: 1133-1141.
- L Zhou, JE Lopes, MM Chong, Ivanov, II, R Min, GD Victora, et al. TGFbeta- induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008; 453: 236-240.
- L Zhou, Ivanov, II, R Spolski R Min, K Shenderov, T Egawa, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007; 8: 967-974.
- MO Li, RA Flavell. TGF-beta: a master of all T cell trades. Cell. 2008; 134: 392-404.
- Gutcher, MK Donkor, Q Ma, AY Rudensky, RA Flavell, MO Li. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 2011; 34: 396-408.
- Ivanov, II, BS McKenzie, L Zhou, CE Tadokoro, A Lepelley, JJ Lafaille, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006; 126: 1121-1133.
- K Ghoreschi, A Laurence, XP Yang, CM Tato, MJ McGeachy, JE Konkel, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signaling. Nature. 2010; 467: 967-971.
- Y Chung, SH Chang, GJ Martinez, XO Yang, R Nurieva, HS Kang, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009; 30: 576-587.
- T Korn, E Bettelli, W Gao, A Awasthi, A Jager, TB Strom, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007; 448: 484-487.
- J Bystrom, FI Clanchy, TE Taher, P Mangat, AS Jawad, RO Williams, et al. TNFalpha in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine. 2018; 101: 4-13.
- S Sakaguchi, N Sakaguchi, M Asano, M Itoh, M Toda. Immunologic selftolerance maintained by activated T cells expressing IL-2 receptor alphachains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995; 155: 1151-1164.
- JD Fontenot, MA Gavin, AY Rudensky. Pillars Article: Foxp3 Programs the Development and Function of CD4+ CD25+ Regulatory T Cells. Nat. Immunol. 2003; 4: 330-336, J Immunol. 2017; 198: 986-992.
- S Hori, T Nomura, S Sakaguchi. Pillars Article: Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science. 2003; 299: 1057- 1061, J Immunol. 2017; 198: 981-985.
- MA Burchill, J Yang, C Vogtenhuber, BR Blazar, MA Farrar. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007; 178: 280-290.
- SZ Josefowicz, LF Lu, AY Rudensky. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012; 30: 531-564.
- B Salomon, DJ Lenschow, L Rhee, N Ashourian, B Singh, A Sharpe, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+ CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000; 12: 431-440.
- TR Malek, A Yu, V Vincek, P Scibelli, L Kong. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002; 17: 167-178.
- S Paust, L Lu, N McCarty, H Cantor. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc Natl Acad Sci USA. 2004; 101: 10398-10403.
- R Pacholczyk, H Ignatowicz, P Kraj, L Ignatowicz. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006; 25: 249-259.
- J Wong, D Mathis, C Benoist. TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J Exp Med. 2007; 204: 2039-2045.
- MA Gavin, TR Torgerson, E Houston, P DeRoos, WY Ho, A Stray-Pedersen, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci USA. 2006; 103: 6659-6664.
- RK Selvaraj, TL Geiger. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J Immunol. 2007; 178: 7667-7677.
- TS Davidson, RJ DiPaolo, J Andersson, EM Shevach. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007; 178: 4022-4026.
- Laurence, CM Tato, TS Davidson, Y Kanno, Z Chen, Z Yao, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007; 26: 371-381.
- J Wang, TW Huizinga, RE Toes. De novo generation and enhanced suppression of human CD4+CD25+ regulatory T cells by retinoic acid. J Immunol. 2009; 183: 4119-4126.
- MF Bachmann, G Kohler, B Ecabert, TW Mak, M Kopf. Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol. 1999; 163: 1128-1131.
- JJ Kobie, PR Shah, L Yang, JA Rebhahn, DJ Fowell, TR Mosmann. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol. 2006; 177: 6780-6786.
- P Pandiyan, L Zheng, S Ishihara, J Reed, MJ Lenardo. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007; 8: 1353-1362.
- S Deaglio, KM Dwyer, W Gao, D Friedman, A Usheva, A Erat, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007; 204: 1257-1265.
- X Yu, K Harden, LC Gonzalez, M Francesco, E Chiang, B Irving, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009; 10: 48-57.
- CL Maynard, RD Hatton, WS Helms, JR Oliver, CB Stephensen, CT Weaver. Contrasting roles for all-trans retinoic acid in TGF-beta-mediated induction of Foxp3 and Il10 genes in developing regulatory T cells. J Exp Med. 2009; 206: 343-357.
- CJ Malemud. Defective T-Cell Apoptosis and T-Regulatory Cell Dysfunction in Rheumatoid Arthritis. Cells. 2018; 7.
- F Safari, S Farajnia, M Arya, H Zarredar, A Nasrolahi. CRISPR and personalized Treg therapy: new insights into the treatment of rheumatoid arthritis. Immunopharmacol Immunotoxicol. 2018; 40: 201-211.
- GA Schellekens, BA de Jong, FH van den Hoogen, LB van de Putte, WJ van Venrooij. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998; 101: 273-281.
- V Baud, M Karin. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001; 11: 372-377.
- WP Arend, JM Dayer. Inhibition of the production and effects of interleukin-1 and tumor necrosis factor alpha in rheumatoid arthritis. Arthritis Rheum. 1995; 38: 151-160.
- LA Joosten, MM Helsen, FA van de Loo, WB van den Berg. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice. A comparative study using anti-TNF alpha, anti-IL-1 alpha/beta, and IL-1Ra. Arthritis Rheum. 1996; 39: 797-809.
- M Veldhoen, RJ Hocking, CJ Atkins, RM Locksley, B Stockinger. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006; 24: 179-189.
- VA Danis, M Millington, V Hyland, R Lawford, Q Huang, D Grennan. Increased frequency of the uncommon allele of a tumour necrosis factor alpha gene polymorphism in rheumatoid arthritis and systemic lupus erythematosus. Dis Markers. 1995; 12: 127-133.
- S Shafia, FA Sofi, Dilafroze, R Rasool, S Javeed, ZA Shah. The association between TNFalpha gene polymorphisms and susceptibility to rheumatoid arthritis in an ethnic Kashmiri population: relationship with disease activity and severity markers. Int J Rheum Dis. 2016; 19: 362-369.
- Finnegan, K Mikecz, P Tao, TT Glant. Proteoglycan (aggrecan)-induced arthritis in BALB/c mice is a Th1-type disease regulated by Th2 cytokines. J Immunol. 1999; 163: 5383-5390.
- P Isomaki, J Punnonen. Pro- and anti-inflammatory cytokines in rheumatoid arthritis. Ann Med. 1997; 29: 499-507.
- C Dong. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008; 8: 337-348.
- HK Park, SK Kim, HY Kweon, KG Lee, MV Arasu, YO Kim. Promoter polymorphism (-590, T/C) of interleukin 4 (IL4) gene is associated with rheumatoid arthritis: An updated meta-analysis. Saudi J Biol Sci. 2017; 24: 444-449.
- SR Harrison, D Li, LE Jeffery, K Raza, M Hewison. Vitamin D, Autoimmune Disease and Rheumatoid Arthritis. Calcif Tissue Int. 2019.
- N Luque-Campos, RA Contreras-Lopez, M Jose Paredes-Martinez, MJ Torres, S Bahraoui, M Wei, et al. Mesenchymal Stem Cells Improve Rheumatoid Arthritis Progression by Controlling Memory T Cell Response. Front Immunol. 2019; 10: 798.
- J Van Snick. Interleukin-6: an overview. Annu Rev Immunol. 1990; 8: 253- 278.
- PC Heinrich, I Behrmann, S Haan, HM Hermanns, G Muller-Newen, F Schaper. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003; 374: 1-20.
- S Rose-John. IL-6 trans signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. 2012; 8: 1237-1247.
- K Schinnerling, JC Aguillon, D Catalan, L Soto. The role of interleukin-6 signalling and its therapeutic blockage in skewing the T cell balance in rheumatoid arthritis. Clin Exp Immunol. 2017; 189: 12-20.
- H Mollazadeh, AFG Cicero, CN Blesso, M Pirro, M Majeed, A Sahebkar. Immune modulation by curcumin: The role of interleukin-10. Crit Rev Food Sci Nutr. 2019; 59: 89-101.
- MJ McGeachy, KS Bak-Jensen, Y Chen, CM Tato, W Blumenschein, T McClanahan, et al. TGF-beta and IL-6 drive the production of IL-17 and IL- 10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007; 8: 1390-1397.
- V Moulin, F Andris, K Thielemans, C Maliszewski, J Urbain, M Moser. B-lymphocytes regulate dendritic cell (DC) function in vivo: increased interleukin 12 production by DCs from B cell-deficient mice results in T helper cell type 1 deviation. J Exp Med. 2000; 192: 475-482.
- M Yang, J Deng, Y Liu, KH Ko, X Wang, Z Jiao, et al. IL-10-producing regulatory B10 cells ameliorate collagen-induced arthritis via suppressing Th17 cell generation. Am J Pathol. 2012; 180: 2375-2385.
- F Apparailly, C Verwaerde, C Jacquet, C Auriault, J Sany, C Jorgensen. Adenovirus-mediated transfer of viral IL-10 gene inhibits murine collageninduced arthritis. J Immunol. 1998; 160: 5213-5220.
- RE Callard, DJ Matthews, L Hibbert. IL-4 and IL-13 receptors: are they one and the same? Immunol Today. 1996; 17: 108-110.
- P Isomaki, R Luukkainen, P Toivanen, J Punnonen. The presence of interleukin-13 in rheumatoid synovium and its antiinflammatory effects on synovial fluid macrophages from patients with rheumatoid arthritis. Arthritis Rheum. 1996; 39: 1693-1702.
- N Bessis, MC Boissier, P Ferrara, T Blankenstein, D Fradelizi, C Fournier. Attenuation of collagen-induced arthritis in mice by treatment with vector cells engineered to secrete interleukin-13. Eur J Immunol. 1996; 26: 2399- 2403.
- Y Onoe, C Miyaura, T Kaminakayashiki, Y Nagai, K Noguchi, QR Chen, et al. IL-13 and IL-4 inhibit bone resorption by suppressing cyclooxygenase-2- dependent prostaglandin synthesis in osteoblasts. J Immunol. 1996; 156: 758-764.
- Y Iwakura, H Ishigame, S Saijo, S Nakae. Functional specialization of interleukin-17 family members. Immunity. 2011; 34: 149-162.
- G Azizi, F Jadidi-Niaragh, A Mirshafiey. Th17 Cells in Immunopathogenesis and treatment of rheumatoid arthritis. Int J Rheum Dis. 2013; 16: 243-253.
- S Kotake, N Udagawa, N Takahashi, K Matsuzaki, K Itoh, S Ishiyama, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999; 103: 1345-1352.
- M Ziolkowska, A Koc, G Luszczykiewicz, K Ksiezopolska-Pietrzak, E Klimczak, H Chwalinska-Sadowska, et al. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000; 164: 2832-2838.
- S Agarwal, R Misra, A Aggarwal. Interleukin 17 levels are increased in juvenile idiopathic arthritis synovial fluid and induce synovial fibroblasts to produce proinflammatory cytokines and matrix metalloproteinases. J Rheumatol. 2008; 35: 515-519.
- E Lubberts, LA Joosten, FA van de Loo, P Schwarzenberger, J Kolls, WB van den Berg. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res. 2002; 51: 102-104.
- E Lubberts, MI Koenders, B Oppers-Walgreen, L van den Bersselaar, CJ Coenen-de Roo, LA Joosten, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004; 50: 650-659.
- S Nakae, Y Komiyama, A Nambu, K Sudo, M Iwase, I Homma, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002; 17: 375-387.
- DS Mehta, AL Wurster, MJ Grusby. Biology of IL-21 and the IL-21 receptor. Immunol Rev. 2004; 202: 84-95.
- DA Young, M Hegen, HL Ma, MJ Whitters, LM Albert, L Lowe, et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007; 56: 1152-1163.
- JM Coquet, S Chakravarti, MJ Smyth, DI Godfrey. Cutting edge: IL- 21 is not essential for Th17 differentiation or experimental autoimmune encephalomyelitis. J Immunol. 2008; 180: 7097-7101.
- LW Collison, CJ Workman, TT Kuo, K Boyd, Y Wang, KM Vignali, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007; 450: 566-569.
- LW Collison, V Chaturvedi, AL Henderson, PR Giacomin, C Guy, J Bankoti, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2010; 11: 1093-1101.
- Kochetkova, S Golden, K Holderness, G Callis, DW Pascual. IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J Immunol. 2010; 184: 7144- 7153.
- W Niedbala, XQ Wei, B Cai, AJ Hueber, BP Leung, IB McInnes, et al. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol. 2007; 37: 3021-3029.
- LI Sakkas, A Mavropoulos, C Perricone, DP Bogdanos. IL-35: a new immunomodulator in autoimmune rheumatic diseases. Immunol Res. 2018; 66: 305-312.
- FL van de Veerdonk, DM de Graaf, LA Joosten, CA Dinarello. Biology of IL- 38 and its role in disease. Immunol Rev. 2018; 281: 191-196.
- J Mai, H Wang, X Yang. T helper 17 cells interplay with CD4+CD25+highFoxp3+ Tregs in regulation of inflammations and autoimmune diseases. Front Biosci. 2011; 15: 986-1006.
- L Xie, Z Huang, H Li, X Liu, S Zheng, W Su. IL-38: A New Player in Inflammatory Autoimmune Disorders. Biomolecules. 2019; 9.
- M Kurowska-Stolarska, S Alivernini, EG Melchor, A Elmesmari, B Tolusso, C Tange, et al. MicroRNA-34a dependent regulation of AXL controls the activation of dendritic cells in inflammatory arthritis. Nat Commun. 2017; 8: 15877.
- L Dong, X Wang, J Tan, H Li, W Qian, J Chen, et al. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J Cell Mol Med. 2014; 18: 2213-2224.
- F Pan, H Xiang, J Yan, L Hong, L Zhang, Y Liu, et al. Dendritic Cells from Rheumatoid Arthritis Patient Peripheral Blood Induce Th17 Cell Differentiation via miR-363/Integrin alphav/TGF-beta Axis. Scand J Immunol. 2017; 85: 441-449.
- S Jin, H Chen, Y Li, H Zhong, W Sun, J Wang, et al. Maresin 1 improves the Treg/Th17 imbalance in rheumatoid arthritis through miR-21. Ann Rheum Dis. 2018; 77: 1644-1652.
- X Tang, K Yin, H Zhu, J Tian, D Shen, L Yi, et al. Correlation Between the Expression of MicroRNA-301a-3p and the Proportion of Th17 Cells in Patients with Rheumatoid Arthritis. Inflammation. 2016; 39: 759-767.
- YH Wu, W Liu, B Xue, L Zhang, XY Liu, B Liu, et al. Upregulated Expression of microRNA-16 Correlates with Th17/Treg Cell Imbalance in Patients with Rheumatoid Arthritis. DNA Cell Biol. 2016; 35: 853-860.
- HY Xiang, F Pan, JZ Yan, LQ Hong, LH Zhang, YH Liu, et al. [Upregulation of miR-498 suppresses Th17 cell differentiation by targeting STAT3 in rheumatoid arthritis patients]. Sheng Li Xue Bao. 2018; 70: 167-174.
- L Yan, M Liang, X Hou, Y Zhang, H Zhang, Z Guo, et al. The role of microRNA-16 in the pathogenesis of autoimmune diseases: A comprehensive review, Biomed Pharmacother. 2019; 112: 108583.
- LF Lu, MP Boldin, A Chaudhry, LL Lin, KD Taganov, T Hanada, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010; 142: 914-929.
- M Dominguez-Villar, CM Baecher-Allan, DA Hafler. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med. 2011; 17: 673-675.
- Q Zhou, S Haupt, JT Kreuzer, A Hammitzsch, F Proft, C Neumann, et al. Decreased expression of miR-146a and miR-155 contributes to an abnormal Treg phenotype in patients with rheumatoid arthritis. Ann Rheum Dis. 2015; 74: 1265-1274.
- HJ Koenen, RL Smeets, PM Vink, E van Rijssen, AM Boots, I Joosten. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008; 112: 2340-2352.
- D Spoerl, I Duroux-Richard, P Louis-Plence, C Jorgensen. The role of miR- 155 in regulatory T cells and rheumatoid arthritis. Clin Immunol. 2013; 148: 56-65.
- S Bluml, M Bonelli, B Niederreiter, A Puchner, G Mayr, S Hayer, et al. Essential role of microRNA-155 in the pathogenesis of autoimmune arthritis in mice. Arthritis Rheum. 2011; 63: 1281-1288.
- M Kurowska-Stolarska, S Alivernini, LE Ballantine, DL Asquith, NL Millar, DS Gilchrist, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci USA. 2011; 108: 11193-11198.
- S Alivernini, M Kurowska-Stolarska, B Tolusso, R Benvenuto, A Elmesmari, S Canestri, et al. MicroRNA-155 influences B-cell function through PU.1 in rheumatoid arthritis. Nat Commun. 2016; 7: 12970.
- M Rajasekhar, AM Olsson, KJ Steel, M Georgouli, U Ranasinghe, C Brender Read, et al. MicroRNA-155 contributes to enhanced resistance to apoptosis in monocytes from patients with rheumatoid arthritis. J Autoimmun. 2017; 79: 53-62.
- Elmesmari, AR Fraser, C Wood, D Gilchrist, D Vaughan, L Stewart, et al. MicroRNA-155 regulates monocyte chemokine and chemokine receptor expression in Rheumatoid Arthritis. Rheumatology (Oxford). 2016; 55: 2056- 2065.
- BK Hong, S You, SA Yoo, D Park, D Hwang, CS Cho, et al. MicroRNA-143 and -145 modulate the phenotype of synovial fibroblasts in rheumatoid arthritis. Exp Mol Med. 2017; 49: e363.