
Mini Review
Austin J Mol & Cell Biol. 2015; 2(1): 1005.
Entrance of Electrons to Acyl-CoA Desaturation
Csala M*
Department of Medical Chemistry, Molecular Biology and Pathobiochemistry, Semmelweis University, Hungary
*Corresponding author: Csala M, Department of Medical Chemistry, Molecular Biology and Pathobiochemistry, Semmelweis University, H-1444 Budapest, POB 260, Hungary
Received: October 04, 2015; Accepted: October 26, 2015; Published: October 28, 2015
Abstract
Deleterious effects of excessive Free Fatty Acid (FFA) supply have been widely studied and are referred to as lipotoxicity. FFA-induced signaling alterations and complex metabolic stress contribute to the increased incidence of various pathologies (e.g. diabetes, cardiovascular diseases and cancer) in obese people. Unsaturated fatty acids are less toxic, and they can reduce the damage caused by saturated ones, and this highlights acyl-CoA desaturation as a cellular defense mechanism. The rate limiting Stearoyl-CoA Desaturase 1 (SCD1) is embedded in the Endoplasmic Reticulum (ER) membrane, and it was known to receive electrons from cytosolic NADH and NADPH. The novel NADH Cytochrome b5 Oxidoreductase (Ncb5or) is assumed to serve as an alternative electron supplier of acyl-CoA desaturases in various tissues. The protein interactions and exact metabolic function of Ncb5or remain to be elucidated. Since the separate pyridine nucleotide pool of the ER lumen is a major determinant of pre-receptor glucocorticoid activation, it’s possible connection with acyl-CoA desaturation is of great importance.
Keywords: Saturated fatty acid; Endoplasmic reticulum; Diabetes; Stress; Insulin resistance; Beta cell dysfunction
Abbreviations
ATF6: Activating Transcription Factor 6; b5: Cytochrome b5; b5R: Cytochrome b5 Reductase; FFA: Free Fatty Acid; H6PD: Hexose 6-Phosphate Dehydrogenase; 11βHSD1: Type 1 11β-Hydroxysteroid Dehydrogenase; IRE1: Inositol-Requiring Enzyme 1; JNK: c-Jun Amino-terminal Kinase; MAPK: Mitogen-Activated Protein Kinase; Ncb5or: NADH Cytochrome b5 Oxidoreductase; PERK RNADependent Protein Kinase-Like ER Kinase; PKC: Protein Kinase C; ROS: Reactive Oxygen Species; SCD1: Stearoyl-CoA Desaturase 1; SFA: Saturated Fatty Acid; UFA: Unsaturated fatty acid; UPR: Unfolded Protein Response
Introduction
Fatty acids are superb sources of metabolic energy in most aerobic cells of the human body. Some tissues, such as the brain have restricted access to plasma lipids due to the presence of special barriers, but others can receive these nutrients either from plasma lipoproteins (i.e. VLDL and Chylomicron) or directly as non-esterified Free Fatty Acids (FFAs). FFAs are largely associated to serum albumin and they are normally available in prolonged starvation (or physical activity) when the fatty acid stores are mobilized and triglycerides are intensively hydrolyzed in the adipocytes [1]. Fat mobilization and FFA secretion are stimulated by low insulin/glucagon ratio and by certain stress hormones, e.g. glucocorticoids and catecholamines. Elevation of FFA levels, therefore, does not normally coincide with insulin action. Obesity and a consequent local inflammation in the adipose tissue can lead to a sustained plasma free fatty academia, which is implicated in the development of diabetes [2] as well as in the increased risk to certain types of cancer [3] observed in obese patients.
Lipotoxicity
Excessive supply of FFA causes a stress of multiple components in the cells of most non-adipose tissues. The damage can result in cellular dysfunction or even in programmed cell death, i.e. lipotoxicity or lipoapoptosis [4]. Upon reaching the outer surface of the plasma membrane, fatty acid molecules can modulate inflammatory signaling through binding to Toll-like receptors [5]. Once taken up into the cytosol, fatty acids are activated to acyl-CoA, an intermediate readily available for synthetic or catabolic purposes. Intensified β-oxidation and coupled oxidative phosphorylation are accompanied by enhanced Reactive Oxygen Species (ROS) generation, which underlies the FFAinduced oxidative stress [6].
Lipotoxicity also involves derangements in the Endoplasmic Reticulum (ER). Permeability of the ER membrane is remarkably increased by long chain acyl-CoA-s at higher concentrations [7], and this causes a disturbance in the luminal milieu [8], primarily in the calcium homeostasis of the organelle. Ca2+ leak interferes with microsomal protein processing as many of the ER chaperones and foldases are calcium-dependent [9]. The consequent accumulation of immature polypeptides in the lumen is sensed by three transmembrane ER stress receptors: RNA-dependent Protein Kinase-like ER Kinase (PERK), Inositol-Requiring Enzyme 1 (IRE1) and Activating Transcription Factor 6 (ATF6). The three largely interacting signaling pathways triggered by these sensors govern major rearrangements in the cellular functions and they are collectively referred to as the Unfolded Protein Response (UPR). Although the primary aim of the UPR is to restore the balance of protein load and protein folding in the ER, it also contributes to insulin resistance and apoptosis [10]. The role of lipotoxicity-induced ER stress in β-cell dysfunction has also been revealed, and attenuation of the UPR has been implicated in the β-cell-protective effect of the antidiabetic drug metformin [11].
Oxidative and ER stress as well as pro-inflammatory stimuli synergistically activate certain members of the Mitogen Activated Protein Kinase (MAPK) family, particularly the c-Jun Amino- Terminal Kinase (JNK). Lipotoxic JNK activation is further supported by diglyceride and ceramide accumulation through atypical Protein Kinase C (PKC) isoforms [12]. The central role of JNK in obesityrelated insulin resistance and diabetes [13] as well as in tumor development and progression [14] has been widely investigated.
Protective role of Fatty Acid Desaturation
It has been demonstrated by several in vivo and in vitro studies that saturated fatty acids (SFAs, e.g., palmitate and stearate) are more toxic than the mono- or poly-unsaturated ones (UFAs, e.g. oleate or linoleate). Moreover, the simultaneously administered UFAs can reduce SFA-induced dysfunctions and injuries. This intriguing phenomenon is partly due to the dominant pro-inflammatory nature of SFAs and anti-inflammatory actions of UFAs, especially n-3 polyunsaturated ones [15]. It became also evident that UFAs are required for an efficient channeling of SFAs to triglyceride synthesis, which serves as a defense mechanism to prevent a deleterious accumulation of saturated acyl-CoA intermediates [4,16]. In addition, IRE1 and PERK can be activated by increased lipid saturation in the ER membrane. Therefore, the ratio of saturated and unsaturated fatty acids in the cell can modulate the UPR signaling through the lipid sensitivity of the transmembrane domains of these receptors [17].
In case of unbalanced supply of SFAs, the intrinsic ability of the cells to insert double bonds into the fatty acyl chains remains the only means to produce protective UFAs and maintain triglyceride synthesis. Human cells employ Stearoyl-CoA Desaturase 1 (SCD1) enzyme to convert stearoyl-CoA (18:0) or palmitoyl-CoA (16:0) to Δ9 mono-unsaturated derivatives, oleyl-CoA (18:1 n9) or palmioleyl- CoA (16:1 n9), respectively [18]. This enzyme is embedded in the ER membrane and receives electrons from NADH or NADPH through the concerted action of two associated membrane proteins, the flavoprotein cytochrome b5 Reductase (b5R) and the hemoprotein cytochrome b5 (b5) (Figure 1). The protective role of SCD1 has been demonstrated in different models of palmitate-induced lipotoxicity [19,20].
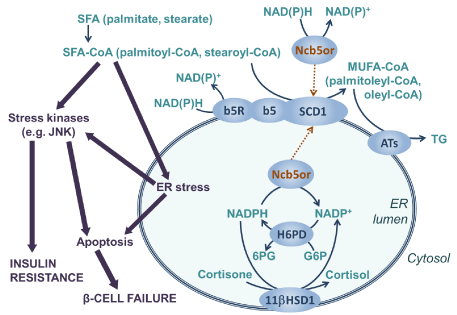
Figure 1: Saturated fatty acid lipotoxicity and microsomal acyl-CoA desaturation.
Saturated Fatty Acids (SFAs) are activated to SFA-CoA, which are largely responsible for the metabolic and signaling stress that leads to insulin resistance and cell
death. Conversion of SFA-CoA to Mono-Unsaturated derivatives (MUFA-CoA) allows Triglyceride (TG) synthesis by Acyltransferases (ATs), and hence decreases
lipotoxicity. Stearoyl-CoA Desaturase 1 (SCD1) can receive electrons from cytosolic NADH or NADPH through cytochrome b5 reductase (b5R) and cytochrome
b5 (b5) in the Endoplasmic Reticulum (ER) membrane. Particulate NADH cytochrome b5 oxidoreductase (Ncb5or) has been postulated as an alternative electron
supplier, and it potentially interacts with the ER luminal glucocorticoid production. Luminal NADPH is produced by Hexose 6-Phosphate Dehydrogenase (H6PD),
which converts Glucose 6-Phosphate (G6P) to 6-Phosphogluconate (6PG). This NADPH is used by type 1 11β-Hydroxysteroid Dehydrogenase (11βHSD1) to
reduce cortisone to cortisol. Solid thin arrows indicate metabolic conversions; solid thick arrows show effects and mechanisms involved in lipotoxicity; Dotted thin
arrows represent the two possible orientations of Ncb5 or-mediated electron transfer in acyl-CoA desaturation. “NAD(P)+” denotes NAD+ or NADP+; “NAD(P)H”
indicates NADH or NADPH.
A recently discovered natural fusion protein, NADH cytochrome b5 oxidoreductase (Ncb5or) has been implicated as an alternative electron supplier of SCD1 or its isoforms [21]. Ncb5or is present in most investigated tissues in mice with the highest expression levels in the pancreas, heart and kidney. Nevertheless, remarkable amounts of the protein have been demonstrated in liver, muscle and brain, too [22]. This soluble enzyme contains both b5R-like and b5-like domains [23], which indicate its participation in fatty acid desaturation. In agreement with this assumption, Ncb5or knock-out mice present with a progressive loss of white adipose tissue and a remarkably reduced ratio of Δ9 desaturated to saturated fatty acids [24]. It is especially intriguing that these animals develop diabetes due to a progressive loss of pancreatic β-cells. It remains to be clarified whether the increased sensitivity of Ncb5or-deficient cells to lipotoxicity [25] is due to a damaged fatty acid unsaturating capacity or it is related to other, yet unrevealed functions of the enzyme.
Redox Connections of Fatty Acid Desaturation
The ER membrane is a barrier between two separated pyridine nucleotide pools [26], i.e. the transmembrane traffic of oxidized NAD+ and NADP+ or reduced NADH and NADPH is too slow to keep pace with the related dehydrogenase activities [8], and hence the states of NAD+-NADH and NADP+-NADPH redox systems are determined by local factors on either side of the ER membrane [27]. Cytosolic NADH predominantly fuels mitochondrial oxidative phosphorylation, and it is produced by various NAD+-dependent dehydrogenases (e.g. glyceraldehyde 3-phosphate dehydrogenase, glycerol dehydrogenase and lactate dehydrogenase). Cytosolic NADPH, in turn, is utilized by major reductive biosynthetic processes (e.g. fatty acid synthesis, fatty acid elongation and cholesterol synthesis), by antioxidant defense mechanisms (e.g. maintenance of reduced glutathione levels) as well as by cytochrome P450 monooxygenases involved in biotransformation, and its supply is maintained by robust mechanisms involving the pentose phosphate pathway, the cytosolic isocitrate dehydrogenase and malic enzyme [28]. The membrane topology of b5R (and b5) makes it evident that the classic electron transfer chain of fatty acyl-CoA desaturation consumes cytosolic NADH and NADPH [29,30].
However, the particulate Ncb5or protein is regarded as a microsomal enzyme, which implies its ER luminal localization [31]. This raises the possibility that SCD1 might receive electrons from the inner NADH/NADPH pool of the organelle. Little is known about the state and function of the ER-luminal NAD+-NADH redox pair. However, it has been long known that local NADP+-NADPH redox cycling couples the activities of two dehydrogenases in this compartment (Figure 1). Local NADPH supply is maintained by Hexose 6-Phosphate Dehydrogenase (H6PD) [32], and it drives the formation of cortisol from cortisone by type 1 11β-Hydroxysteroid Dehydrogenase (11βHSD1) in many tissues (e.g. liver, muscle, adipose tissue) [33]. This redox machinery is capable of pre-receptor glucocorticoid production, and hence alterations in the ER luminal NADP+-NADPH redox state has an impact on the cortisol action in the given cells and tissues [34]. Oxidation of ER luminal NADPH by metyrapone has been shown to prevent cortisone-induced adipocyte differentiation [35], and a similar mechanism has been also implicated in the anti-diabetic activity of epigallocatechin gallate [36]. The possibility that Ncb5or may link this redox system to acyl-CoA desaturation is of great importance because it could serve as a novel mechanism of nutrient sensing [37], and it would likely be involved in the development of lipotoxicity.
Conclusion
Cellular fatty acyl-CoA pool can be increased by import across the plasma membrane, by cytosolic synthesis and by intracellular hydrolysis of triglycerides while it can be decreased by mitochondrial β-oxidation and by incorporation into triglycerides and various membrane lipids. Excessive supply of free fatty acids causes a metabolic and signaling stress called lipotoxicity, which is partly due to the accumulation of acyl-CoA and a consequently enhanced β-oxidation. Since the protective triglyceride synthesis requires a proportional supply of unsaturated acyl-CoAs, it largely depends on the capacity of microsomal desaturation systems.
Although the participation of Ncb5or in the microsomal fatty acyl-CoA desaturation has not yet been proven directly, its domain structure and the phenotype of Ncb5or-/- mice strongly support the assumption that this novel oxidoreductase can deliver electrons from NADH and NADPH to SCD1. This possibility is intriguing even if Ncb5or channels the cytosolic reducing power in the unsaturating machinery, as it is done by the classic electron transfer chain composed of b5R and b5. However, the proposed emplacement of the soluble Ncb5or in the ER suggests that a direct redox link might exist between the luminal pre-receptor cortisol activation and acyl- CoA desaturation. In this case Ncb5or deficiency would not only impair a protective mechanism that reduces the saturated acyl-CoA burden but also enhance the local glucocorticoid producing capacity of 11βHSD1, which is regarded as a pro-diabetic factor [38].
The human relevance of Ncb5or is highlighted by the existence of natural mutations that destabilize the protein structure and reduce the cellular level of the human enzyme through accelerated proteasomal degradation [39]. Subcellular localization, redox connections and functions of this novel oxidoreductase, therefore, deserve further investigation, which might yield a better understanding of lipotoxicity, and ER redox homeostasis, too.
Acknowledgement
The author thanks the Hungarian Scientific Research Fund (OTKA 106060) for its support.
References
- Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by longchain fatty acids. Prog Lipid Res. 2014; 53: 124-144.
- Reaven GM, Chen YD. Role of abnormal free fatty acid metabolism in the development of non-insulin-dependent diabetes mellitus. Am J Med. 1988; 85: 106-112.
- Gong Y, Dou LJ, Liang J. Link between obesity and cancer: role of triglyceride/ free fatty acid cycling. Eur Rev Med Pharmacol Sci. 2014; 18: 2808-2820.
- Zámbó V, Simon-Szabó L, Szelényi P, Kereszturi E, Bánhegyi G, Csala M. Lipotoxicity in the liver. World J Hepatol. 2013; 5: 550-557.
- Yin J, Peng Y, Wu J, Wang Y, Yao L. Toll-like receptor 2/4 links to free fatty acid-induced inflammation and β-cell dysfunction. J Leukoc Biol. 2014; 95: 47-52.
- Seifert EL, Estey C, Xuan JY, Harper ME. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during longchain fatty acid oxidation. J Biol Chem. 2010; 285: 5748-5758.
- Bánhegyi G, Csala M, Mandl J, Burchell A, Burchell B, Marcolongo P, et al. Fatty acyl-CoA esters and the permeability of rat liver microsomal vesicles. Biochem J. 1996; 320: 343-344.
- Csala M, Marcolongo P, Lizák B, Senesi S, Margittai E, Fulceri R, et al. Transport and transporters in the endoplasmic reticulum. Biochim Biophys Acta. 2007; 1768: 1325-1341.
- Csala M, Kereszturi é, Mandl J, Bánhegyi G. The endoplasmic reticulum as the extracellular space inside the cell: role in protein folding and glycosylation. Antioxid Redox Signal. 2012; 16: 1100-1108.
- Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes (Lond). 2008; 32: S52-54.
- Simon-Szabo L, Kokas M, Mandl J, Keri G, Csala M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS One. 2014; 9: e97868.
- Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, et al. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. 2004; 18: 2024-2034.
- Li H, Yu X. Emerging role of JNK in insulin resistance. Curr Diabetes Rev. 2013; 9: 422-428.
- Bubici C, Papa S. JNK signalling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol. 2014; 171: 24-37.
- Kennedy A, Martinez K, Chuang CC, LaPoint K, McIntosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. J Nutr. 2009; 139: 1-4.
- Cheon HG, Cho YS. Protection of palmitic acid-mediated lipotoxicity by arachidonic acid via channeling of palmitic acid into triglycerides in C2C12. J Biomed Sci. 2014; 21: 13.
- Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA. 2013; 110: 4628-4633.
- Miyazaki M, Ntambi JM. Role of stearoyl-coenzyme a desaturase in lipid metabolism. Prostaglandins Leukot Essent Fatty Acids. 2003; 68: 113-121.
- Peter A, Weigert C, Staiger H, Machicao F, Schick F, Machann J, et al. Individual stearoyl-coa desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is associated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes. 2009; 58: 1757-1765.
- Green CD, Olson LK. Modulation of palmitate-induced endoplasmic reticulum stress and apoptosis in pancreatic β-cells by stearoyl-CoA desaturase and Elovl6. Am J Physiol Endocrinol Metab. 2011; 300: E640-649.
- Zhu H, Qiu H, Yoon HW, Huang S, Bunn HF. Identification of a cytochrome b-type NAD(P)H oxidoreductase ubiquitously expressed in human cells. Proc Natl Acad Sci USA. 1999; 96: 14742-14747.
- Larade K, Bunn HF. Promoter characterization and transcriptional regulation of Ncb5or, a novel reductase necessary for pancreatic beta-cell maintenance. Biochim Biophys Acta. 2006; 1759: 257-262.
- Deng B, Parthasarathy S, Wang W, Gibney BR, Battaile KP, Lovell S, et al. Study of the individual cytochrome b5 and cytochrome b5 reductase domains of Ncb5or reveals a unique heme pocket and a possible role of the CS domain. J Biol Chem. 2010; 285: 30181-30191.
- Larade K, Jiang Z, Zhang Y, Wang W, Bonner-Weir S, Zhu H, et al. Loss of Ncb5or results in impaired fatty acid desaturation, lipoatrophy, and diabetes. J Biol Chem. 2008; 283: 29285-29291.
- Zhang Y, Larade K, Jiang ZG, Ito S, Wang W, Zhu H, et al. The flavoheme reductase Ncb5or protects cells against endoplasmic reticulum stressinduced lipotoxicity. J Lipid Res. 2010; 51: 53-62.
- Csala M, Bánhegyi G, Benedetti A. Endoplasmic reticulum: a metabolic compartment. FEBS Lett. 2006; 580: 2160-2165.
- Piccirella S, Czegle I, Lizak B, Margittai E, Senesi S, Papp E, et al. Uncoupled redox systems in the lumen of the endoplasmic reticulum. Pyridine nucleotides stay reduced in an oxidative environment. J Biol Chem. 2006; 281: 4671-4677.
- Rzezniczak TZ, Merritt TJ. Interactions of NADP-reducing enzymes across varying environmental conditions: a model of biological complexity. G3 (Bethesda). 2012; 2: 1613-1623.
- Oshino N, Imai Y, Sato R. A function of cytochrome b5 in fatty acid desaturation by rat liver microsomes. J Biochem. 1971; 69: 155-167.
- Ozols J. The role of microsomal cytochrome b5 in the metabolism of ethanol, drugs and the desaturation of fatty acids. Ann Clin Res. 1976; 8: 182-192.
- Zhu H, Larade K, Jackson TA, Xie J, Ladoux A, Acker H, et al. NCB5OR is a novel soluble NAD(P)H reductase localized in the endoplasmic reticulum. J Biol Chem. 2004; 279: 30316-30325.
- Senesi S, Csala M, Marcolongo P, Fulceri R, Mandl J, Banhegyi G, et al. Hexose-6-phosphate dehydrogenase in the endoplasmic reticulum. Biol Chem. 2010; 391: 1-8.
- Czegle I, Piccirella S, Senesi S, Csala M, Mandl J, Banhegyi G, et al. Cooperativity between 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase is based on a common pyridine nucleotide pool in the lumen of the endoplasmic reticulum. Mol Cell Endocrinol. 2006; 248: 24-25.
- Bánhegyi G, Csala M, Benedetti A. Hexose-6-phosphate dehydrogenase: linking endocrinology and metabolism in the endoplasmic reticulum. J Mol Endocrinol. 2009; 42: 283-289.
- Marcolongo P, Senesi S, Gava B, Fulceri R, Sorrentino V, Margittai E, et al. Metyrapone prevents cortisone-induced preadipocyte differentiation by depleting luminal NADPH of the endoplasmic reticulum. Biochem Pharmacol. 2008; 76: 382-390.
- Szelenyi P, Revesz K, Konta L, Tutto A, Mandl J, Kereszturi E, et al. Inhibition of microsomal cortisol production by (-)-epigallocatechin-3-gallate through a redox shift in the endoplasmic reticulum--a potential new target for treating obesity-related diseases. Biofactors. 2013; 39: 534-541.
- Mandl J, Mészáros T, Bánhegyi G, Hunyady L, Csala M. Endoplasmic reticulum: nutrient sensor in physiology and pathology. Trends Endocrinol Metab. 2009; 20: 194-201.
- Csala M, Margittai E, Bánhegyi G. Redox control of endoplasmic reticulum function. Antioxid Redox Signal. 2010; 13: 77-108.
- Kálmán FS, Lizák B, Nagy SK, Mészáros T, Zámbó V, Mandl J, et al. Natural mutations lead to enhanced proteasomal degradation of human Ncb5or, a novel flavoheme reductase. Biochimie. 2013; 95: 1403-1410.