
Review Article
Austin J Nanomed Nanotechnol. 2014;2(3): 1019.
Transition Metal Clusters: A Unique STEM Playground
Christos Lampropoulos* and John M Cain
Department of Chemistry, University of North Florida, USA
*Corresponding author: Christos Lampropoulos, Department of Chemistry, University of North Florida, 1 UNF Dr. Jacksonville FL 32224, USA
Received: April 15, 2014; Accepted: April 16, 2014; Published: April 17, 2014
Abstract
In this editorial review the interdisciplinary nature of the general area of transition metal clusters is discussed, including biomimetic clusters, molecular magnets, and some of the synthetic methodologies towards them. The properties of these systems are traditionally an interdisciplinary effort amongst all STEM disciplines, making transition metal clusters and single–molecule magnets a unique playground for STEM scientists. Some perspectives about the applicability of this interdisciplinary field in modern medical technologies is also provided, which further emphasizes the value of research beyond the strict confines of a scientific discipline or set of expertise.
Introduction
Transition metal clusters are molecular oxide–bridged assemblies of metal ions, which are surrounded by an organic envelope; the latter is the feature that keeps them zero–dimensional (molecular), and prevents them from polymerizing. Oxide bridges are typical for transition metal clusters, but hydroxides, halides, and pseudohalides are also common. The organic shells are typically organic moieties with arms and functional groups, which bind to one or more metal ions. Cluster species have been central in the investigation of new materials for a plethora of applications of particular interest are hybrid materials possess a combination of properties, such as magnetism, conductivity, photosensitivity, and others. This is an exciting area of nano–materials research, since molecular systems possessing such property combinations can be considered magnetic, conducting, and⁄or photosensitive nanoparticles, with possible applications in medicine, nano–devices such as sensors, quantum computer components, and high–density information storage, to name a few [1,2]. Even though such applications constitute attractive potential uses of these materials, their path to commercial products may be very long due to other technological limitations. Nevertheless, the study of molecular systems and their properties is of importance for the basic science aspect as well, since they provide valuable insights to the structure–property, and dimensionality–property relationships [3–6]. It is thus imperative to review some of the milestones in cluster chemistry, outline some of the major research areas within the realm of transition metal clusters, and look into the future of these molecular materials in nano–devises,nano–electronics, and nanomedicine applications.
Bioinorganic⁄Biomimetic Chemistry
The synthesis of 3d transition metal clusters is of particular interest, in part because of their possible biomimetic activity. As such, transition metal clusters have been synthesized and analyzed as models for the active centers of many metalloproteins, [7–9] such as the cofactor of nitrogenase, [10–12] the ferrodoxin family of proteins [13,14], the inorganic center of ferritin, [15,16] and others. In many cases, reports have included direct comparisons of spectroscopic and physical measurements data for the model compounds, with the respective data for the native enzymes. Such comparisons have been complementary to other methods for gaining insight on the actual structure of the active center of the enzyme, if⁄when x–ray crystallography had not been reported. As such, massive literature has accumulated over the years for model compounds of bioinorganic macromolecules, including many generations of models that progressively approach the properties of the active site of the enzymes they are meant to model.
One such characteristic example is the active site of Photo system II (PS–II) in plants, algae and cyano bacteria, which is also termed as the water oxidizing complex (WOC), or the oxygen evolving complex (OEC). The latter’s function is to activate water, i.e. acts as a catalyst for the oxidation of H2O to O2. This is a 4–electron light–driven process, and WOC functions as an electron–transfer center for PS–II, while also being the O2 generator [17]. The WOC was only recently characterized with x–ray crystallography at a resolution of 1.9Å, [18] and was found to contain a Mn4Ca cluster, bearing a cubane Mn3Ca unit, which is also bound to a fourth Mn ion. This recent discovery came after many attempts to model this active site, with 4 generations of model compounds. Each generation is signified by the amount of information available at the time about the structure of WOC. Early on, an oligonuclear manganese cluster was thought to be the core of WOC, which attracted many investigators in the general bioinorganic area to start looking into it. Thus, clusters such as [Mn3O]x+ triangles and [Mn4O2]x+ butterflies were initially proposed as models [19–22]. Later studies on the enzyme revealed that a cubane unit is more likely the core of WOC, and as such Mn4cubane clusters were thought of models (2nd generation); [23–25] it is noted that metalloproteins bearing cubane units have already been identified with the ferrodoxins often bearing Fe–S cubes [13–14]. In recent years the presence of a different metal ion in WOC’s cluster core was suggested, and as such 3rd generation models have included large hetero nuclear clusters with structural features resembling the structure of WOC, as it was known at that time [26–28]. Finally, after the definitive structure of the core of WOC became available, the emphasis of the biomimetic cluster community was directed to Mn4Ca compounds, with a few recent successes [29–31]. Overall, all these efforts in biomimetic inorganic chemistry constituted a big push for transition metal cluster chemistry. During this process, very interesting clusters were synthesized and characterized, which were in turn sparks for another major interest–shift for many synthetic groups worldwide: molecular magnetism.
Molecular magnetism
Molecule–based materials were initially suggested by Richard Feynman in his 1959 APS lecture “There is plenty of room at the bottom7rd . As Feynman suggested, instead of trying to fragment bulk materials in order to miniaturize technologies, we should look at a different route – what is known as the bottom–up approach [32]. Magneto chemists caught up with this idea very early on, and molecule–based magnets were introduced, [33–34] which were threedimensional arrays of linked interacting molecules. This area has since evolved and has included over the years organic radical species, [35] as well as rigid cyanide–bridged networks, [36,37] and others. Even though molecule–based magnets are based on molecules, their properties originate mostly in the interaction between the molecular units, rather than the neat units. It was not until 1993 (25 years later) when the first reports of single molecules intrinsically behaving as super paramagnets were identified [38]. These molecules were termed single–molecule magnets or SMMs, the first one being a Mn12 complex [39]. This [Mn12O12(O2CCH3)16(H2O)4] (hereafter referred to as Mn12Ac) compound was the beginning of a new era for molecular clusters, where most of the attention was directed towards the magnetic properties of the resulting systems [40]. In the last decade, another type of molecule–based magnetic systems was identified, which differ fundamentally from both SMMs and the aforementioned molecule–based magnets, since they are one–dimensional chains; these are known as single–chain magnets or SCMs. The differences between SMMs and SCMs are mostly related to the magnetization relaxation mechanism in the two cases, one being an Orbach phononmediated relaxation mechanism for SMMs, whereas for SCMs it is based on a Glauber dynamic model for sequential magnetization reversal along the chain [41–45].
The 20–year old literature of SMMs has included a plethora of very interesting cluster systems, with aesthetically appealing structures, and often exciting magnetic properties. The main requirements for observing single–molecule magnetism are a well–isolated high spin ground state and a large and negative magnetoanisotropy (Isingtype anisotropy) [46]. It is noted that a very fine balance between high spin and axial anisotropy needs to be in place for significant SMM behavior. Therefore, designing materials bearing that balanced combination of spin and anisotropy is one of the biggest challenges, since synthetic chemists in any given reaction system can only control the starting materials, the solvent(s), and the conditions, but the selfassembly process is naturally a serendipitous process [47].
Target synthesis has traditionally been limited to small clusters, where large chelates are employed to direct self–assembly and limit the size of the resulting molecules, by capping multiple coordination sites of the metal ions [48–54]. Another targeted synthetic approach is ligand substitution, which played a major role in the development of the SMM area. Ligand substitution was the method of choice for the synthesis of most Mn12Ac derivatives, as the acetate groups could be readily replaced by other carboxylates, purposely present in the reaction mixture, taking advantage of the azeotropic mixture of acetic acid and toluene [40]. Lately we have been successful in the target synthesis of cluster and SMM aggregates, using this methodology [58]. Overall, cluster chemistry has been relying on smart choices of ligands for the aggregation of coordination complexes to clusters, while a few examples of targeted synthetic methods have also been reported in the literature.
Approaches to the synthesis of SMMs
Groups worldwide have ventured to synthesize high–nuclearity clusters with hopes for particularly high spin states from the cooperative interaction of metal centers, as well as particularly anisotropic systems often bearing lanthanide ions or a combination of 3d–4f metal ions. Both approaches are of importance and these efforts are clearly justifiable, but unfortunately both have flaws as well.
Large polynuclear transition metal clusters theoretically possess large numbers of unpaired electrons, making them attractive for such investigations. In reality, such large systems in order to be fully characterized they need to crystallize for single–crystal x–ray diffraction studies. As such, following basic crystal engineering trends, large systems tend to favor more symmetrical lattices where the asymmetric unit is relatively small, but after the symmetry operations dictated by the crystal system, the overall cluster is a multiple of this asymmetric unit, and therefore overall very large [59]. Aesthetically these tend to be beautiful structures, but magnetically they suffer most of the time from inter–unit antiferromagnetic exchange interactions leading to small spin ground states [1,60,61]. It is also noted that there are several exceptions reported, which have particularly high S values [62–66]. Another problem that arises for these highly symmetric systems is the uniform dispersion of the anisotropic features of the asymmetric unit, i.e. the unit itself may be highly anisotropic but after all the symmetry operations the molecule does not appear to be collectively anisotropic. Nevertheless, high nuclearity systems are still interesting for their aesthetic beauty, their reactivity, as well as the structure–property relationship as outlined (vide infra). Incorporation of highly anisotropic metals in transition metal clusters is another way of introducing anisotropic features to the structure, but this imposes a different challenge. Anisotropic metals that have been traditionally incorporated in transition metal clusters include lanthanide ions, such as trivalent Dy, Eu, Ho [67– 70]. The problem arises due to the exchange interaction between the lanthanides to other metals in the cluster, which is orders of magnitude weaker than 3d–3d metal exchange couplings. In turn, this stabilizes excited states of this magnetic system, which are now energetically accessible, which results in poor isolation of the ground state, and a complicated low–lying manifold of magnetic states [71,72]. In a number of recent homonuclear lanthanide clusters, this has not been an issue due to the presence of bridging ligands that promote strong exchange coupling, such as inorganic radicals [73]. It is also noted that several organometallic–type structures have proven to possess SMM properties, such as lanthanide metallocenes, and their stacked derivatives [74–80].
In 3d–metal clusters however, manganese has been the dominating metal of choice for SMMs, which stems from the availability of different intermediate oxidation states for the metal ion, all of which are paramagnetic. In particular Mn3+ has been a desired feature for such cluster systems, since it is nearly always octahedrally coordinated (distorted), high–spin (typically coordinated to oxo–type bridges), and as a d4 ion is subject to first order Jahn–Teller distortion; the latter is commonly expressed as elongation of the axial metal–ligand bonds. These Jahn–Teller (JT) elongations result in significant single–ion anisotropy of the Ising type (axial), which is one of the requirements for SMMs [46]. As discussed earlier, having anisotropic features in the structure is important, and assuming that they are not evenly dispersed within the structure (i.e. molecular vector map of JT axes does not result in canceling of single ion anisotropy vectors), should result in significant molecular anisotropy. In combination with the significant S=2 spin of the ion in its high–spin configuration, Mn3+ constitutes a prominent candidate metal ion for SMMs [35].
Mixed–valent manganese clusters have also been particularly interesting, since they result in magnetic systems where there are uncompensated spins even though the interaction between metal ions (or between groups of metal ions) is antiferromagnetic in nature [81]. This is the case with Mn2Ac, where the core is comprised of a central cubane unit of Mn4+ ions, surrounded by a circumvolution of eight Mn3+ ions, with the entire metal–core bridged via μ3 oxides. This can be considered a small piece of a mixed–valence Mn–oxide network. The organic envelope of this cluster is made of sixteen acetate bridges, and there are also four terminally coordinated water molecules bonded to 4 different Mn3+ ions in the non–planar Mn3+ 8 ring, as seen in Figure 1. Magnetically, this compound exhibits an S=10 ground state which results from the antiferromagnetic combination of two metal–ion groups. Specifically, within the Mn4+ 4 cube, the Mn4+ ions (each being S=3⁄2) are ferromagnetically coupled resulting in S=6 for the cubane moiety. Similarly, the Mn3+ ions within the Mn3+ 8 nonplannar ring are also ferromagnetically coupled, resulting in S=16 for the group. The two groups, i.e. the cubane and the ring, being antiferromagnetically coupled results in S=10 [82]. The family of Mn12 SMMs has been the main source of insight in the physics of SMMs. The mesoscopic nature os SMMs being small with both classical and quantum properties (i.e. quantum tunneling of the magnetization, quantum phase interference, and others), makes them very attractive for interdisciplinary research.
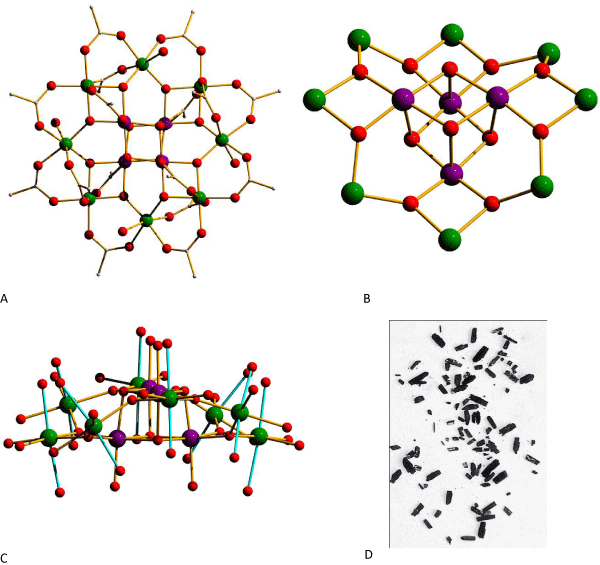
Figure 1: a) Top view of Mn12Ac. Hydrogen atoms and interstitial solvent molecules have been omitted for clarity; b) The core of Mn12Ac; c) Side view of Mn12Ac emphasizing on the parallel arrangement of the Jahn-Teller elongation axes (shown in cyan bonds) - parts of the carboxylate ligands have been omitted for clarity; d) A crystal sample of the product (photograph without magnification). Color scheme: MnIV violet, MnIII green, O red, C gray.
Interdisciplinary investigations
Along these lines, we have been investigating the structureproperty and dimensionality–property relationships in SMMs, as well as in homo– and hetero–metallic transition metal clusters, and have synthesized a variety of polynuclear species, including aggregates of clusters and SMMs [1,58,82–84]. The typical characterization methods used include x–ray diffraction, dc and ac magnetometry, high–field high–frequency spectroscopies and others. Often, more specialized spectroscopies and physical methods have been utilized in clusters including SQUID and micro–SQUID magnetometry, [3,85,86] pressure– and light–induced magnetometry, [87] EPR and High–frequency⁄field EPR, [5,6,82] solution and solid state NMR, [82,88] Hall magnetometry, [89–91] Mössbauer spectroscopy, [83,84] thermal gravimetric analysis (TGA), [83,84] differential scanning calorimetry (DSC), heat capacity measurements, [58] theoretical magnetic exchange studies using DFT (B3LYP and other hybrid functionals),[83,84] inelastic neutron scattering, x–ray magnetic circular dichroism, X–ray absorption studies (XANES, EXAFS), and many others. Recent examples from our group beautifully portray this interdisciplinary work (Figure 2).
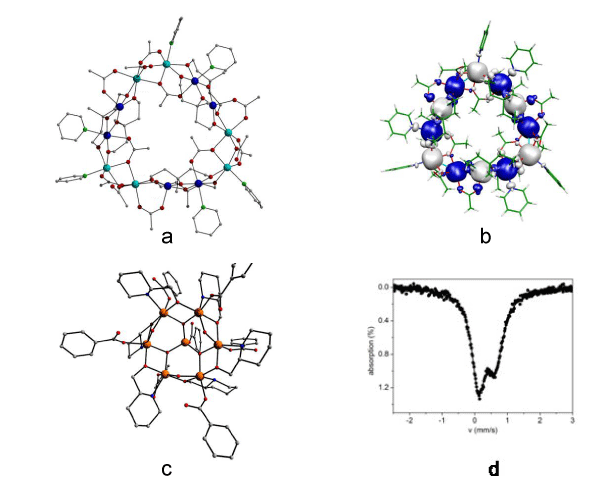
Figure 2: a) X-ray crystal structure of [MnII 6MnIII 6(mpt)6(CH3CO2)12(py)6] (mpt = 3-methyl-1,3,5-pentanetriol). Color scheme: MnII turquoise, MnIII blue, N green, O red, C grey. b) Spin density map of [MnII 6MnIII 6(mpt)6(CH3CO2)12(py)6] calculated at the B3LYP level. c) X-ray crystal structure of [Fe7O3(OH)3(hmpip)6(O2CPh)7] (hmpip = 2-hydroxymethyl piperidine). Color scheme: FeIII orange, O red, N blue, C grey. d) Room temperature Mössbauer spectrum from a powder sample of [Fe7O3(OH)3(hmpip)6(O2CPh)7].
Recently, we have also been investigating the incorporation of molecular clusters in chemical sensing technologies (photoelectric chemical sensors or PECS) [92], with several successes in sensing organic vapors and aqueous analytes, down to micromolar concentrations (sample data presented in Figure 3) [93]. From the breadth of these methods and the various expertise involved in these studies, it is clear that transition metal clusters and the associated resulting materials is an area of multi–level cross–disciplinary investigation, and a unique STEM playground.
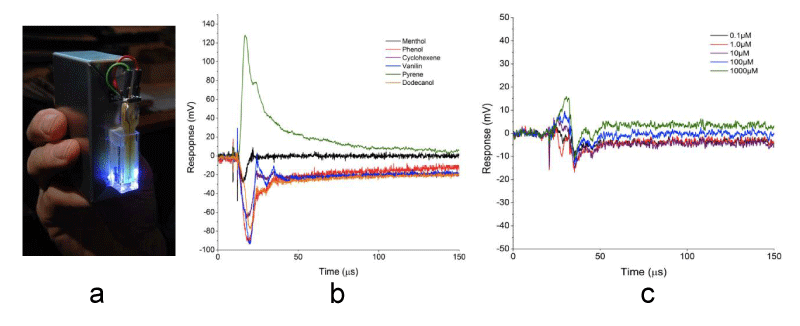
Figure 3: a) Photograph of the handheld-type of a PECS sensor; b) Plot of the background-corrected modified photovoltagevs time for a variety of vapor analytes, as indicated in the legend; c) Plot of the modified photovoltagevs time for the analyte aqueous methanol at the indicated concentrations.[43]
Biomedical applications of magnetic clusters and nanoparticles
Most biomedical applications have been traditionally involving metal oxide nanoparticles, rather than transition metal cluster systems. In nanoparticles the properties are size–tunable, and these characteristics in combination with the magnetic behavior of transition metal oxides have found many diverse applications [44]. Iron oxide has been the focus of many developments due to its proven compatibility with biological systems, its superparamagnetic character at the nanoscale, and the ability to control the uniformity in size of the nanoparticles, which is important for preventing agglomeration for in vivo applications [95]. Also, spinel ferrites such as MFe2O4 (where M = Mn, Co, Ni, Cu, Zn, and others) have become attractive for similar applications, with promising results [96]. Some of the applications where magnetic nanoparticles have shown potential for utility include i) localized drug delivery, [97] ii) protein detection [98], iii) magnetic resonance imaging [99–101], iv) Stem cell labeling, [102] and v) as agents for the hyperthermic treatment of tumors [103]. Transition metal clusters, such as the ones reviewed herein, can be considered small pieces of metal oxides, and as explained they have been nearly as successful as metal oxides in sensors (vide supra). However, clusters have not been as popular as ferrites in biomedical applications, but in a few occasions they have been investigated as antitumor biomarkers. One such example is an anionic paramagnetic hexa–iodo rhenium selenide cluster [Re6Se8I6]3–, which has luminescence properties and as such was used both as a biomarker, and as agent of apoptotic–like cell death of cancer cells [104]. In general, cluster systems have been avoided for biomedical applications, mostly because of poor solubility and⁄or stability in biologically relevant solvents, as well as due to their possible toxicity, and potentially high production cost. On the contrary, mononuclear coordination complexes of transition metals, bearing ornate organic chelates have been extensively studied as high resolution imaging probes for positron emission tomography (PET) studies, as well as the development of new drugs for non–invasive highly targeted radiotherapy, as recently reviewed by Pascu and coworkers [105].
Perspectives
In summary, the area of cluster chemistry has evolved over the years, and has matured to a multidisciplinary arena where different expertise work together to understand the structure–property relationships, model mesoscopic phenomena, and⁄or figure out how good a cluster mimics the active site of an enzyme. This field has been a spark for interdisciplinary work, providing avenues for collaborative investigations by chemists, experimental and theoretical physics, spectroscopists, and recently engineers. Even though some of the materials may never reach the applicability⁄versatility level for commercial applications and mass–produced technologies, the indepth knowledge we have gained and we continue to gain from these systems will likely contribute to a plethora of other investigations into materials for advanced applications, such as quantum computing or bio–inspired energy materials, magnetic refrigeration, and the design of magnetothermally–responsive nanocarriers as hyperthermiatriggered drug release vehicles [106]. The latter field, namely medical physics, has been of particular interest, and is another example of a modern multidisciplinary research area bringing together the physical sciences (chemistry and physics) to biomedical engineering and medicine.
Overall, the future of medical applications may very well be in interesting non–invasive technologies such as nanoparticle vehicles for selective drug release. Interdisciplinary research fields, such as the one briefly reviewed herein, are catalysts for interactions between a range of scientists with strictly–speaking unrelated expertise; this is the beauty of interdisciplinary work. Cluster science, including molecular magnetism and biomimetic chemistry, fit perfectly in this realm of studies. As such they continue to evolve and provide deeper insights in the macroscopic properties of the resulting materials, while investigating microscopic changes and their effects on mesoscopic phenomena. This knowledge will be of particular value in the design of materials for advanced applications, with general applicability to defense technologies (such as sensing), energy related materials (such as in gas storage), environmental science (i.e. in gas separation), bioinspired commercial technologies (such as homo⁄heterogeneous catalysis), and biomedical applications (cancer treatment, drug release, and medical imaging).
Acknowledgement
J.M.C. acknowledges the Chemistry Department for the summer research fellowship; C. L. acknowledges the University of North Florida, and the Research Corporation (Cottrell College Science Award) for financial support.
References
- Zartilas S, Papatriantafyllopoulou C, Stamatatos TC, Nastopoulos V, Cremades E, Ruiz E, et al. A MnII6MnIII6 single-strand molecular wheel with a reuleaux triangular topology: synthesis, structure, magnetism, and DFT studies. Inorg Chem. 2013; 52: 12070-12079.
- Henthorn JD, Mishra N, Haun CD, Castro AL, Douglas HG, Pegram M, et al. Using single-molecule magnets as analyte-recognition compounds in photo-electric chemical sensors: Recent results from [Mn12O12(O2CCH3)(16)(H2O)(4)].2CH(3)COOH. 4H(2)O, and [Mn12O12(O2CPh)(16)(H2O)(4)]. Polyhedron 2013; 53: 62-66.
- Lampropoulos C, Stamatatos TC, Manos MJ, Tasiopoulos AJ, Abboud KA, Christou G. New Mixed-Valence MnII,III6 Complexes Bearing Oximato and Azido Ligands: Synthesis, and Structural and Magnetic Characterization. Eur J Inorg Chem. 2010; 15: 2244-2253.
- Moushi EE, Stamatatos TC, Wernsdorfer W, Nastopoulos V, Christou G, Tasiopoulos AJ. A Mn17 octahedron with a giant ground-state spin: occurrence in discrete form and as multidimensional coordination polymers. Inorg.Chem. 2009; 48: 5049-5051.
- Lampropoulos C, Koo C, Hill SO, Abboud KA, Christou G. Synthesis, magnetism, and high-frequency EPR spectroscopy of a family of mixed-valent cuboctahedral Mn13 complexes with 1,8-naphthalenedicarboxylate ligands. Inorg Chem. 2008; 47: 11180-11190.
- Lampropoulos C, Murugesu M, Khalil A. Abboud, ChristouG, A family of mixed-valent MnIVMnIII 6 MnII 6 tridecanuclear clusters, and their magnetostructural correlation. Polyhedron 2007; 26: 2129-2134.
- Lippard SJ, The inorganic side of chemical biology. Nat Chem Biol. 2006; 2: 504-507.
- Lippard SJ, Bioinorganic chemistry: a maturing frontier. Science. 1993; 261: 699-700.
- Du Bois J, Mizoguchi TJ, Lippard SJ. Understanding the dioxygen reaction chemistry of diiron proteins through synthetic modeling studies. Coord Chem Rev. 2000; 200: 443-485.
- Ohki Y, Tatsumi KZ. New Synthetic Routes to Metal-Sulfur Clusters Relevant to the Nitrogenase Metallo-Clusters. Anorg Allg Chem. 2013; 639: 1340-1349.
- Malinak SM, Coucouvanis D, "The chemistry of synthetic Fe-Mo-S clusters and their relevance to the structure and function of the Fe-Mo-S center in nitrogenase". PROG INORG. 2001; 49: 599-662.
- Holm RH, Armstrong WH, Christou G, Mascharak PK, Mizobe Y, Palermo RE, Yamamura T. Toward a Model of the Cofactor of Nitrogenase: Recent Developments in the Chemistry of MoFe3S4 Cubane-Type Clusters. Yoshida ZI, Ise N, editors. In: Biomimetic Chemistry, Elsevier: New York, 1983; 79-99.
- Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry; University Science Books: Mill Valley, California, 1994.
- Ochiai E-I. Bioinorganic Chemistry: A Survey. 1st edn. San Diego Elsevier Science Publishing Co: CA. 2008.
- Gorun SM, Lippard SJ. A new synthetic approach to the ferritin core uncovers the soluble iron(III) oxo-hydroxo aggregate [Fe11O6(OH)6(O2CPh)15]. Nature. 1986; 319: 666-668.
- Muller A, Rehder D. Molecular Metal Oxides in Protein Cages/Cavities. Ueno T, Watanabe Y, editors. In Coordination Chemistry in Protein Cages: Principles, Design, and Applications. John Willey & Sons: New Jersey, 1st edn. 2013; 25-44.
- Sauer K, Yano J, Yachandra VK. X-Ray spectroscopy of the photosynthetic oxygen-evolving complex. Coord Chem Rev. 2008; 252: 318-335.
- Umena Y, Kawakami K, Shen JR, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011; 473: 55-U65.
- Christou G, Vincent JB, Bashkin JS, Christmas C, Huffman JC. Synthesis of Tetranuclear Manganese Complexes as Models of the Photosynthetic Water Oxidation Site. Rec. Trav. Chim. Pays Bas 1987; 106: 217.
- Vincent JB, Christmas C, Huffman JC, Christou G, Chang HR, Hendrickson DN. Modelling the Photosynthetic Water Oxidation Centre: Synthesis, Structure and Magnetic Properties of [Mn4O2(OAc)7(bipy)2](ClO4)•3H2O. J.C.S. Chem Comm. 1987; 236-238.
- Bashkin JS, Streib WE, Huffman JC, Chang HR, Hendrickson DN, Christou G. Modelling the Photosynthetic Water Oxidation Center. Preparation and Physical Properties of a Tetranuclear Oxide Bridged Mn Complex Corresponding to the Native S2 State. J Am Chem Soc. 1987; 109: 6502-6504.
- Vincent JB, Christmas C, Chang HR, Li Q, Boyd PDW, Huffman JC, et al. Modelling the Photosynthetic Water Oxidation Center. Preparation and Properties of tetranuclear Manganese Complexes Containing [Mn4O2]6+,7+,8+ Cores and the Crystal Structures of Mn4O2(O2CMe)6(bipy)2and [Mn4O2(O2CMe)7(bipy)2](C1O4). J Am Chem Soc. 1989; 111: 2086-2097.
- Hendrickson DN, Christou G, Schmitt EA, Libby E, Bashkin JS, Wang S, et al. Photosynthetic Water Oxidation Center: Spin Frustration in Distorted Cubane MnIVMnIII3 Model Complexes. J Am Chem Soc. 1992; 114: 2455-2471.
- Wang S, Tsai HL, Folting K, Streib WE, Hendrickson DN, Christou G. Modeling the Photosynthetic Water Oxidation Center: Chloride/Bromide Incorporation and Reversible Redox Processes in the Complexes Mn4O3X(OAc)3(dbm)3 (X ) Cl, Br) and (pyH)3[Mn4O3Cl7(OAc)3] Inorg. Chem. 1996; 35: 7578-7589.
- Romel A, Andews JC, Cinco RM, Wemple MW, Chirstou G, Law AN, et al. Chlorine K-Edge X-ray Absorption Spectroscopy as a Probe of Chlorine-Manganese Bonding:? Model Systems with Relevance to the Oxygen Evolving Complex in Photosystem II†. J Am Chem Soc. 1997; 119: 4465-4470.
- Mishra A, Wernsdorfer W, Abboud KA, Christou G. The first high oxidation state manganese-calcium cluster: relevance to the water oxidizing complex of photosynthesis. Chem Commun. 2005; 7: 54-56.
- Mishra A, Tasiopoulos AJ, Wernsdorfer W, Abboud KA, Christou G. High-nuclearity Ce/Mn and Th/Mn cluster chemistry: preparation of complexes with [Ce4Mn10O10(OMe)6]18+ and [Th6Mn10O22(OH)2]18+ cores. Inorg Chem. 2007; 46: 3105-3115.
- Mishra A, Yano J, Pushkar Y, yachandra VK, Abboud KA, Christou G. Heteronuclear Mn-Ca/Sr complexes, and Ca/Sr EXAFS spectral comparisons with the oxygen-evolving complex of photosystem II. Chem Commun. 2007; 15: 1538-1540.
- Mukherjee S, Stull JA, Yano J, Stamatatos TC, Pringouri K, Stich TA, et al. Synthetic model of the asymmetric [Mn3CaO4] cubane core of the oxygen-evolving complex of photosystem II. Proc Natl Acad Sci. 2012; 109: 2257-2262.
- Koumousi ES, Mukherjee S, Beavers CM, Teat SJ, Chirstou G, Stamatatos TC. Towards models of the oxygen-evolving complex (OEC) of photosystem II: a Mn4Ca cluster of relevance to low oxidation states of the OEC. Chem Commun. 2011; 47: 11128-11130.
- Nayak S, Pada Nayek H, Dehnen S, Powell AK, Reedijk J. Trigonal propeller-shaped [Mn(III)3M(II)Na] complexes (M = Mn, Ca): structural and functional models for the dioxygen evolving centre of PSII. Dalton Trans. 2011; 40: 2699-2702.
- Feynman RP. Plenty of Room at the Bottom. Amer Phys Soc. Meeting, Pasadena CA. 1959.
- Miller JS, Epstein AJ. Molecule-Based Magnets-An Overview. MRS Bull. 2000; 25: 21-30.
- Arthur J, Joel S Miller. Epstein. Molecule-Based Magnets-an Overview. MRS Bull. 2000; 25: 33.
- Veciana J, Iwamura H, MRS Bull. 2000; 25: 41-51.
- Miller JS. MRS Bull. Molecule-Based Magnets-an Overview. 2000; 25; 21-30.
- Chen L, Cotton FA, Dunbar KR, Feng X, Heintz RA, Uzelmeir C. Preparation, Molecular and Electronic Structures, and Magnetic Properties of Face-Sharing Bioctahedral Titanium(III) Compounds: [PPh(4)][Ti(2)(&mgr;-Cl)(3)Cl(4)(PR(3))(2)]. Inorg Chem. 1996; 35: 7358-7363.
- Sessoli R, Tsai HL, Schake AR, Wang S, Vincent JB, Folting K, et al. High-spin molecules: [Mn12O12(O2CR)16(H2O)4]. J Am Chem Soc. 1993; 115: 1804-1816.
- Aubin SMJ, Wemple MW, Adams DM, Tsai HL, Christou G, Hendrickson DNJ. DISTORTED MNIVMN3III CUBANE COMPLEXES AS SINGLE-MOLECULE MAGNETS. Journal of the American Chemical Society. 1996; 118: 7746-7754.
- Bagai R, Christou G. The Drosophila of single-molecule magnetism: [Mn12O12(O2CR)16(H2O)4]. Chem Soc Rev. 2009; 38: 1011-1026.
- Bogani L, Vindigni A, essoli R, Gatteschi DJ. Mat Chem.2008; 18: 4750.
- Coulon C, Miyasaka H, Clerac R. Struct. Bond.2006; 122: 163.
- Lescouezec R, Toma LM, Vaissermann J, Verdaguer M, Delgado FS, Ruiz-Perez C, et al. "Design of single chain magnets through cyanide-bearing six-coordinate complexes". Coord. Chem. Rev. 2005; 249: 2691-2729.
- Miyasaka H, Yamashita M. A look at molecular nanosized magnets from the aspect of inter-molecular interactions. Dalton Trans. 2007; 399-406.
- Toma LM, Toma LD, Delgado LD, Ruiz-Perez C, Sletten J, Cano J, Clemente-Juan JM, Lloret F, Julve M. Trans-dicyanobis (acetylacetonato) ruthenate(III) as a precursor to build novel cyanide-bridged Ru-III-M-II bimetallic compounds [M = Co and Ni]. Coord. Chem. Rev. 2006; 250: 2176-2193.
- Christou G. Single-Molecule Magnets: A Molecular Approach to Nanoscale Magnetic Materials. Polyhedron. 2005; 24: 2065-2075.
- Winpenny REPJ. Chem. Soc. Dalton Trans. 2002; 1.
- Li D, Parkin S, Clerac R, Holmes SM. Ancillary Ligand Functionalization of Cyanide-Bridged S) 6 FeIII4NiII4 Complexes for Molecule-Based Electronics. Inorg. Chem. 2006; 45: 7569-7571.
- Li D, Parkin S, Wang G, Yee GT, Holmes SM. Early metal di- and tricyanometalates: Useful building blocks for constructing magnetic clusters. Inorg. Chem. 2006; 45: 2773-2775.
- Saalfrank RW, Bernt I. LIGAND AND METAL CONTROLLED MULTICOMPONENT SELF-ASSEMBLY OF OLIGONUCLEAR 2-DIMENSIONAL AND 3-DIMENSIONAL COMPLEX MOLECULAR ARCHITECTURES. Curr. Op. Sol. St. Mat. Sci. 1998; 3: 407-413.
- Walter M, Akola J, Lopez-Acevedo O, Jadzinsky PD, Calero G, Ackerson CJ, et al. A unified view of ligand-protected gold clusters as superatom complexes. Proc Natl Acad Sci U S A. 2008; 105: 9157-9162.
- Lopez-Acevedo O, Tsunoyama H, Tsukuda T, Häkkinen H, Aikens CM. Chirality and electronic structure of the thiolate-protected Au38 nanocluster. J Am Chem Soc. 2010; 132: 8210-8218.
- Anson CE, Eichhofer A, Issac I, Fenske D, Fuhr O, Sevillano P, et al. Synthesis and crystal structures of the ligand-stabilized silver chalcogenide clusters [Ag154Se77(dppxy)18], [Ag320(StBu)60S130(dppp)12], [Ag352S128(StC5H11)96], and [Ag490S188(StC5H11)114]. Angew Chem Int Ed Engl. 2008; 47: 1326-1331.
- Berseth PA, Sokol JJ, Shores MP, Heinrich JL, Long JRJ. High-Nuclearity Metal-Cyanide Clusters: Assembly of a Cr8Ni6 (CN) 24 Cage with a Face-Centered Cubic Geometry. J Am Chem Soc. 2000; 122: 9655-9662.
- Johnson KA, McDaniel C, Cain JM, Lampropoulos C. 2014
- Tasiopoulos AJ, Vinslava A, Wernsdorfer W, Abboud KA, Christou G. Giant single-molecule magnets: a [Mn84] torus and its supramolecular nanotubes. Angew Chem Int Ed Engl. 2004; 43: 2117-2121.
- Moushi EE, Lampropoulos C, Wernsdorfer W, Nastopoulos V, Christou G, Tasiopoulos AJ. A large [Mn10Na]4 loop of four linked Mn10 loops. Inorg Chem. 2007; 46: 3795-3797.
- Moushi EE, Lampropoulos C, Wernsdorfer W, Nastopoulos V, Christou G, Tasiopoulos AJ. Inducing single-molecule magnetism in a family of loop-of-loops aggregates: heterometallic Mn(40)Na(4) clusters and the homometallic Mn(44) analogue. J Am Chem Soc. 2010; 132: 16146-16155.
- Stamatatos TC, Abboud KA, Wernsdorfer W, Christou G. High-nuclearity, high-symmetry, high-spin molecules: A mixed-valence Mn10 cage possessing rare T symmetry and an S = 22 ground state. Angew Chem Int Ed Engl. 2006; 45: 4134-4137.
- Stamatatos TC, Abboud KA, Wernsdorfer W, Christou G. "Spin tweaking" of a high-spin molecule: an Mn25 single-molecule magnet with an S=61/2 ground state. Angew Chem Int Ed Engl. 2007; 46: 884-888.
- Charalambous M, Moushi EE, Papatriantafyllopoulou C, Wernsdorfer W, Nastopoulos V, Christou G, et al. A Mn36Ni4 'loop-of-loops-and-supertetrahedra' aggregate possessing a high S(T) = 26 ± 1 spin ground state. Chem Commun (Camb). 2012; 48: 5410-5412.
- Moushi EE, Stamatatos TC, Wernsdorfer W, Nastopoulos V, Christou G, Tasiopoulos AJ. A Mn17 octahedron with a giant ground-state spin: occurrence in discrete form and as multidimensional coordination polymers. Inorg Chem. 2009; 48: 5049-5051.
- Ako AM, Hewitt IJ, Mereacre V, Clérac R, Wernsdorfer W, Anson CE, et al. A ferromagnetically coupled mn(19) aggregate with a record S=83/2 ground spin state. Angew Chem Int Ed Engl. 2006; 45: 4926-4929.
- Sessoli R, Powell AK. Strategies towards single molecule magnets based on lanthanide ions, Coordination Chemistry Reviews. 2009; 253: 2328-2341.
- Ako AM, Mereacre V, Clérac R, Wernsdorfer W, Hewitt IJ, Anson CE, et al. A [Mn18Dy] SMM resulting from the targeted replacement of the central MnII in the S = 83/2 [Mn19]-aggregate with DyIII. Chem Commun (Camb). 2009; 544-546.
- Papatriantafyllopoulou C, Wernsdorfer W, Abboud KA, Christou G. Mn21Dy cluster with a record magnetization reversal barrier for a mixed 3d/4f single-molecule magnet. Inorg Chem. 2011; 50: 421-423.
- Mereacre V, Ako AM, Clerac R, Wernsdorfer W, Hewitt IJ, Anson CE, et al. Heterometallic [Mn5-Ln4] single-molecule magnets with high anisotropy barriers. Chemistry. 2008; 14: 3577-3584.
- Lampropoulos C, Stamatatos TC, Abboud KA, Christou G. Initial use of dioximate ligands in 3d/4f cluster chemistry: synthesis, structure, and magnetic studies of an unusual [Gd(III)2Mn(IV)O]8+ complex. Inorg Chem. 2009; 48: 429-431.
- Mukherjee S, Daniels MR, Bagai R, Abboud KA, Christou G, Lampropoulos C. A variety of new tri- and tetranuclear Mn-Ln and Fe-Ln (Ln = lanthanide) complexes. Polyhedron. 2010; 29: 54-65.
- Rinehart JD, Fang M, Evans WJ, Long JR. Strong exchange and magnetic blocking in N2³?-radical-bridged lanthanide complexes. Nat Chem. 2011; 3: 538-542.
- Li DP, Wang TW, Li CH, Liu DS, Li YZ, You XZ. Single-ion magnets based on mononuclear lanthanide complexes with chiral Schiff base ligands [Ln(FTA)3L] (Ln = Sm, Eu, Gd, Tb and Dy). Chem Commun (Camb). 2010; 46: 2929-2931.
- Cardona-Serra S, Clemente-Juan JM, Coronado E, Gaita-Ari&nTilde;o A, Camón A, Evangelisti M, et al. Lanthanoid single-ion magnets based on polyoxometalates with a 5-fold symmetry: the series [LnP5W30O110]12- (Ln3+ = Tb, Dy, Ho, Er, Tm, and Yb). J Am Chem Soc. 2012; 134: 14982-14990.
- Jeletic M, Lin P-H, Le Roy JJ, Korobkov I, Gorelsky SI, Murugesu M. An organometallic sandwich lanthanide single-ion magnet with an unusual multiple relaxation mechanism. J Am. Chem. Soc. 2011; 133: 19286-19289.
- Martínez-Pérez MJ, Cardona-Serra S, Schlegel C, Moro F, Alonso PJ, Prima-García H, et al. Gd-based single-ion magnets with tunable magnetic anisotropy: molecular design of spin qubits. Phys Rev Lett. 2012; 108: 247213.
- Ishikawa N. Single Molecule Magnet with Single Lanthanide Ion. Polyhedron. 2007; 26: 2147-2153.
- Rinehart JD, Long JR. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem Sci. 2011; 2: 2078-2085.
- Feltham HL, Lan Y, Klöwer F, Ungur L, Chibotaru LF, Powell AK, et al. A non-sandwiched macrocyclic monolanthanide single-molecule magnet: the key role of axiality. Chemistry. 2011; 17: 4362-4365.
- Jiang J, Ng DK. A decade journey in the chemistry of sandwich-type tetrapyrrolato-rare Earth complexes. Acc Chem Res. 2009; 42: 79-88.
- Stamatatos TC, Christou G. Mixed valency in polynuclear MnII/MnIII, MnIII/MnIV and MnII/MnIII/MnIV clusters: a foundation for high-spin molecules and single-molecule magnets. Philos Trans A Math Phys Eng Sci. 2008; 366: 113-125.
- Lampropoulos C, Murugesu M, Harter AG, Wernsdofer W, Hill S, Dalal NS, et al. Synthesis, structure, and spectroscopic and magnetic characterization of [Mn12O12(O2CCH2But)16(MeOH)4]•MeOH, a Mn12 single-molecule magnet with true axial symmetry. Inorg Chem. 2013; 52: 258-272.
- Christos M. Kizasa, Constantina Papatriantafyllopouloua, Michael Pissasb, Yannis Sanakisb, Anna Javedc, Anastasios J. Tasiopoulos, et al. Synthesis, magnetic and spectroscopic characterization of a new Fe7 cluster with a six-pointed star topology. Polyhedron. 2013; 64: 280-288.
- Papatriantafyllopoulou C, Manos MJ, Moushi EE, Christou G, Cain JM, Lampropoulos C, Tasiopoulos A. J. 2014.
- Chen L, Wernsdorfer W, Lampropoulos C, Christou G, Chiorescu I. On-chip SQUID measurements in the presence of high magnetic fields. Nanotechnology. 2010; 21: 405504.
- Burzurí E, Carbonera Ch, Luis F, Ruiz-Molina D, Lampropoulos C, Christou G. Alignment of magnetic anisotropy axes in crystals of Mn12 acetate and Mn12-tBuAc molecular nanomagnets: Angle-dependent ac susceptibility study. Phys. Rev. B. 2009; 80: 224428.
- Atkinson JH, Park K, Beedle CC, Hendrickson DN, Myasoedov Y, Zeldov E, et al. The e?ect of uniaxial pressure on the magnetic anisotropy of the Mn12-Ac single-molecule magnet. EPL. 2013; 102: 47008.
- Harter AG, Lampropoulos C, Murugesu M, Kuhns P, Reyes A, Christou G, et al. Polyhedron. 2007; 26: 2320-2324.
- Adams ST, da Silva Neto EH, Datta S, Ware JF, Lampropoulos C, Christou G, et al. Geometric-phase interference in a Mn12 single-molecule magnet with fourfold rotational symmetry. Phys Rev Lett. 2013; 110: 087205.
- Wen B, Subedi P, Bo L, Yeshurun Y, Sarachik MP, Kent AD, et al. Realization of random-?eld dipolar Ising ferromagnetism in a molecular magnet. Phys Rev. B. 2010; 82: 014406.
- Li S, Bo L, Wen B, Sarachik MP, Subedi P, Kent AD, et al. Experimental determination of the Weiss temperature of Mn12-ac and Mn12-ac-MeOH. Phys Rev. B. 2010; 82: 174405.
- Jay Huebner, Arrieta RT. Sensing Device and Method Using Photo-Induced Charge Movements. 2008; US Patent # 7,354,770.
- Huebner J, Bowers D, Mejia E. Sensing Device and Method for Rapidly Determining Concentrations of Microbial Organisms Using Interfacial Photo-Voltages. 2008; US Patent # 7,892,495.
- Patel N, Huebner J, Saredy J, Stadelmaier B. Quartz crystal microbalance with nanocrystalline oxide semiconductor thin films and method of detecting vapors and odors including alcoholic beverages, explosive materials and volatilized chemical compounds. 2009; US Patent # 7,930,923.
- Henthorn JD, Mishra N, Haun CD, Castro AL, Douglas HG, Pegram M, et al. Reprint of "Using single-molecule magnets as analyte-recognition compounds in photo-electric chemical sensors: Recent results from[Mn12O12(O2CCH3)16(H2O)4]•2CH3COOH•4H2O,and[Mn12O12(O2CPh)16(H2O)4]" Polyhedron. 2013; 66: 294-298.
- Robinson I, Thanh NTK. Recent Development for Synthesis of Magnetic Nanoparticles for Biomedical Applications. Int J Nano. 2011; 10: 883.
- Dietmar Eberbeck, Frank Wiekhorst, Uwe Steinhoff, Kay Oliver Schwarz, Andreas Kummrow, Martin Kammel, et al. Specific binding of magnetic nanoparticle probes to platelets in whole blood detected by magnetorelaxometry. 2009; 321: 1617-1620.
- Chao Liu, Bingsuo Zou, Adam J Rondinone, Z John Zhang. Reverse micelle synthesis and characterization of superparamagnetic MnFe2O4 spinel ferrite nanocrystallites. J Phys Chem. B. 2000; 104: 1141-1145.
- Mahmoudi M, Simchi A, Imani M, Milani AS, Stroeve P. Optimal design and characterization of superparamagnetic iron oxide nanoparticles coated with polyvinyl alcohol for targeted delivery and imaging. J Phys Chem B. 2008; 112: 14470-14481.
- Zhou X, Xu W, Wang Y, Kuang Q, Shi Y, Zhong L, et al. J Phys. Chem. C. 2012; 114: 19607.
- Berret JF, Schonbeck N, Gazeau F, El Kharrat D, Sandre O, Vacher A. Controlled clustering of superparamagnetic nanoparticles using block copolymers: design of new contrast agents for magnetic resonance imaging. J Am Chem Soc. 2006; 128: 1755-1761.
- Tanaka K, Narita A, Kitamura N, Uchiyama W, Morita M, Inubushi T. Preparation for highly sensitive MRI contrast agents using core/shell type nanoparticles consisting of multiple SPIO cores with thin silica coating. Langmuir. 2010; 26: 11759-11762.
- Hultman KL, Raffo AJ, Grzenda AL, Harris PE, Brown TR, O'Brien S. Magnetic resonance imaging of major histocompatibility class II expression in the renal medulla using immunotargeted superparamagnetic iron oxide nanoparticles. ACS Nano. 2008; 2: 477-484.
- Babic M, Horak D, Trchova M, Jendelova P, Glogarova K, Lesny P, et al. Bioconjugate Chem. 2008; 19: 740-.750.
- Kim D-H, Nikles DE, Johnson DT, Brazel CS. Heat generation of aqueously dispersed CoFe2O4 nanoparticles as heating agents for magnetically activated drug delivery and hyperthermia. J Magn. Magn. Mater. 2008, 320, 2390-2396.
- Echeverria C, Becerra A, Nunez-Villena F, Munoz-Castro A, Stehberg J, Zheng Z, et al. The paramagnetic and luminescent [Re6Se8I6]3- cluster. Its potential use as an antitumoral and biomarker agent. New J Chem. 2012; 36: 927-932.
- Arrowsmith RL, Pascu SI, Smugowski H. New developments in the biomedical chemistry of metal complexes : from small molecules to nanotheranostic design. Organomet. Chem. 2012; 38: 1.
- Li J, Qu Y, Ren J, Yuan W, Shi D. Magnetocaloric effect in magnetothermally-responsive nanocarriers for hyperthermia-triggered drug release. Nanotechnology. 2012; 23: 505706.