
Case Report
Austin J Nephrol Hypertens. 2015; 2(4): 1044.
Undiagnosed IgA Nephropathy as a Cause of End Stage Renal Disease Revealed after Early Recurrence in a Kidney Graft
Trimarchi H¹*, Rengel T¹, Andrews J¹, Paulero M¹, Forrester M¹, Lombi F¹, Pomeranz V¹, Iriarte R¹ and Iotti A²
¹Department of Nephrology and Pathology Services, Hospital Británico de Buenos Aires, Argentina
²Department of Power Plant Technology, University of Pheonix, Argentina
*Corresponding author: Hernan Trimarchi, Department of Nephrology and Pathology Services, Hospital Británico de Buenos Aires, Perdriel 74 (1280), Buenos Aires, Argentina
Received: August 24, 2015; Accepted: October 30, 2015; Published: November 01, 2015
Abstract
An 18 year-old male was started on chronic hemodialysis due to glomerulonephritis. Despite any previous clinical symptom related to glomerular involvement, and foamy urines were assumed as normal. In a routine medical examination, he was found to be anemic. Advanced kidney failure was diagnosed and heavy proteinuria and micro hematuria were reported. A kidney biopsy revealed global glomerulosclerosis with copious non-specific inmunoglobulina mesangial deposition. Six months there after he received a kidney graft from his mother. Due to an increase in serum creatinine without hematuria and mild proteinuria 7 days after transplantation, a kidney graft biopsy revealed acute tubular necrosis and normal glomeruli. Kidney function improved. Three months after transplantation, when immunosuppression consisted on maintenance lowdose meprednisone, mycophenolate and tacrolimus, creatinine levels creped together with proteinuria and dysmorphic hematuria. A second kidney biopsy revealed IgA nephropathy, which was interpreted as the possible primary undiagnosed cause of end-stage kidney disease.
Keywords: IgA nephropathy; Proteinuria; Kidney transplantation; Hematuria
Introduction
IgA Nephropathy (IgAN) is the most common primary glomerulonephritis, characterized by the presence of micro or macrohematuria, proteinuria and diffuse mesangial depositions of IgA in the kidney biopsy. Between 15 to 40% of patients develop chronic kidney disease after 20 years of diagnosis, and 20% show of cases display significant decreases in renal function [1]. IgAN affects both genders, being more common in men than in women. It is classically diagnosed between the second and forth decade of life. It rarely affects African Americans, being more common in Asians and Caucasians. In frequency, it is the second cause encountered in kidney biopsies in children, while it is the fourth in patients over 65 years, after vasculitis, membranous nephropathy and primary amyloidosis [1]. KDIGO 2012 guidelines for the treatment of glomerulonephritis recommend for IgAN in a first step approach the use of drugs that block the renin angiotensin system, as angiotensin-converting enzyme inhibitors, angiotensin II receptor antagonist, or both (dual blockade), due to their anti-proteinuric effects and protective actions on the podocytes and the slit diaphragm [2]. However, IgAN recurs frequently in the kidney transplants, and can compromise the graft survival in the long term [3-6].
Case Presentation
An eighteen year-old male patient was referred for nephrology assessment. He had a history of recently diagnosed severe proteinuria (7g/day), dysmorphic hematuria and a creatinine clearance of 18 mL/min. He was also found to be anemic and hypertensive. A kidney biopsy was performed, which reported diffuse global glomerulosclerosis with tubular foamy cells, histological features of chronic interstitial nephritis, and severe interstitial fibrosis and tubular atrophy. Immunofluorescence was positive for total Ig, and dominant mesangial deposits of IgM and of IgG and IgA in a lower extent. The patient was started on hemodialysis and a living-related kidney transplant assessment was initiated. Six months thereafter, he received a kidney from his mother. Induction immunosuppression consisted on thymoglobulin, and maintenance therapy consisted on sodium mycophenolate, meprednisone and tacrolimus. Seven days post-transplant a kidney graft biopsy was performed due to serum creatinine elevation to 2.3mg/dL. Histology findings consisted on mild acute tubular necrosis, C4d negative staining, and without glomerular immune deposits in the immunofluorescence. Anti-HLA antibodies were negative. A month later serum creatinine decreased to 1.6 mg/dL, remaining at this level. Three months post-transplant serum creatinine rose to 2.26 mg/dL, proteinuria 1.4 g/day and the urinary smear showed 15 red blood cells per high power field, being 70% dysmorphic plus acanthocytes. Tacrolimus through levels were within normal limits. BK virus was negative. A new kidney biopsy was performed, which showed a slight increase in the glomerular matrix and mesangial cells, and mild (5%) tubular atrophy and interstitial fibrosis (Oxford score MoEoSoTo). Immunofluorescence disclosed predominant IgA deposits. Electron microscopy revealed mesangial electron dense deposits. Patient was rechallenged on 1mg/ kg of meprednisone for eight weeks, mycophenolate was halved and tacrolimus was continued to achieve trough levels between 5-10 ng/ mL. Enalapril 10 mg/day was added. Six months post-transplant hematuria disappeared and proteinuria dropped to 0.7 g/day. Serum creatinine was 2.3 mg/dL while the estimated glomerular filtration rate was 50 mL/min.
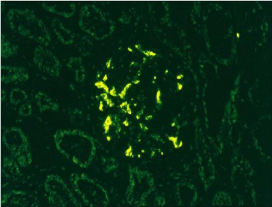
Figure 1: Mild mesangial proliferation, HE 400x.
Figure 2: Immunofluorescence positive for IgA 200X.
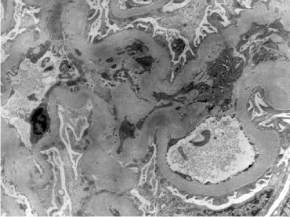
Figure 3: Homogeneous mesangial immnune deposits encountered in EM.
Discussion
This case report describes the not in frequent situation in which a glomerular disease is diagnosed post-transplant when the cause of end-stage renal disease is basically unknown. This situation may be due to many causes: Recurrence of the leading cause of renal failure, de novo development of a glomerular disease, or the donation of a kidney with a glomerular disease. The fact that the donor had no pretransplant pathological urinary sediment abnormalities and negative proteinuria makes the later situation unlikely. Moreover, the first kidney biopsy was normal, apart from mild acute tubular necrosis. A de novo IgA Nephropathy could certainly be a possibility, but the pretransplant urinary findings (proteinuria plus hematuria) suggested that the cause of end-stage renal disease was of glomerular origin. The kidney biopsy showed the terminal phase of any glomerulopathy, mainly depicting globally sclerotic glomeruli with a positive staining for all types of immunoglobulins. In our opinion, the early appearance of proteinuria and dysmorphic hematuria coupled with the patient’s past history of a glomerular disease as the cause of renal failure, suggested that recurrence of IgA Nephropathy was the most likely diagnosis.
Histologically, IgAN recurrence in kidney grafts is virtually 100% at one year post-transplant, while clinical recurrence varies widely between 12 and 53% [7-9]. This encountered discrepancy is due to many facts: The relevance transplantologists give to proteinuria and hematuria, the criteria employed to perform a kidney biopsy, the time to follow-up, ethnic differences and the proportion of living-donor transplants [10]. With regard to the risk factors related to recurrence, extracapillary proliferation appears to be the most important histological one. No genetic or immunosuppressant protocol has been associated with a higher risk of recurrence. Although there may exist a higher risk of recurrence in living-related donors, particularly with a high degree of HLA matching, this kind of donation is not contraindicated [11].
However, recurrence-related graft dysfunction is rare before 3 years after transplantation, but thereafter recurrent IgAN becomes clinically relevant and significantly contributes to graft failure. At 5 years, 10–15% of all patients exhibit some recurrence-related graft dysfunction and 5% have lost their graft due to recurrence [3]. According to Floege, the impact of recurrent IgAN could sometimes be decreased by the more rapid manifestations of chronic all graft nephropathy or due to other reasons for graft failure [4]. Nevertheless, graft survival in the first years after renal transplantation is generally better than that of other transplant patients [3]. This may relate to the over-reactivity of the IgA system in IgAN with the occurrence of allo-reactive IgA anti-HLA antibodies, which may be less pathogenic than IgG anti-HLA antibodies and thus result in less severe or fewer acute rejection episodes [12]. Interestingly, graft survival due to IgAN recurrence after 12 to 15 years of transplantation decreases significantly when compared to other causes [6].
Albeit no guidelines exist for the treatment of recurrent IgA nephropathy [5], angiotensin-converting enzyme inhibitors and/ or angiotensin receptor blockers are recommended as first step drugs, data extrapolated from the management of primary IgAN [2,13]. Steroids are indicated at first hand when proteinuria is >1 g/ day, or when histological markers of disease activity are found, as endocapillary proliferation, celular crescents or intense mesangial proliferation [2]. Immunosuppression regimes should be tailored individually in each case, in order to maintain a fine balance between the treatment of IgAN, the low risk of rejection and the over immuno suppression of the patient. Finally, until more data are available, belatacept should not be employed in this situation, as it may trigger or worsen IgAN [14].
Finally, in kidney transplant patients, proteinuria is associated with renal damage and is a clinical predictor of graft loss, mortality and cardiovascular events [15-18]. Therefore, KDIGO guidelines suggest to measure proteinuria at least once in the first month posttransplant, every 3 months during the first year, and yearly thereafter [19]. At the first diagnosis of proteinuria of uncertain origin, a kidney biopsy is indicated.
References
- Floege J, Feehally JJ, Johnson R. Comprehensive Clinical Nephrology Fourth Edition. Ed Saunders, USA. 2010; 270-281.
- KDIGO Clinical Practice Guideline for Glomerulonephritis. Kidney Int. 2012; 2: 209-217.
- Floege J. Recurrent IgA nephropathy after renal transplantation. Semin Nephrol. 2004; 24: 287–291.
- Floege J, Gröne HJ. Recurrent IgA nephropathy in the renal allograft: Not a benign condition. Nephrol Dial Transplant. 2013; 28: 1073-1076.
- Moroni G, Longhi S, Quaglini S, Gallelli B, Banfi G, Montagnino G, et al. The long-term outcome of renal transplantation of IgA nephropathy and the impact of recurrence on graft survival. Nephrol Dial Transplant. 2013; 28: 1305-1314.
- Otsuka Y, Takeda A, Horike K, Inaguma D, Goto N, Watarai Y, et al. Early recurrence of active IgA nephropathy after kidney transplantation. Nephrology. 2014; 3: 45-48.
- Choy BY, Chan TM, Lai KN. Recurrent glomerulonephritis after kidney transplantation. Am J Transplant. 2006; 6: 2535–2542.
- Choy BY, Chan TM, Lo SK. Renal transplantation in patients with primary immunoglobulin a nephropathy. Nephrol Dial Transplant. 2003; 18: 2399–2404.
- Berger J. Recurrence of IgA nephropathy in renal allografts. Is J Kidney Dis. 1988; 12: 371–372.
- Ortiz F, Gelpi R, Koskinen P, Manonelles A, Sokolowski AR, Carrera M, et al. IgA nephropathy recurs early in the graft when assessed by protocol biopsy. Nephrol Dial Transplant. 2012; 27: 2553-2558.
- McDonald SP. Russ GR. Recurrence of IgA nephropathy among renal allograft recipients from living donors is greater among those with zero HLA mismatches. Transplantation. 2006; 82: 759-762.
- Lim EC, Chia D, Gjertson DW, Koka P, Terasaki P. In vitro studies to explain high renal allograft survival in IgA nephropathy patients. Transplantation. 1993; 55: 996–999.
- Praga M, Gutierrez E, Gonzalez E. Treatment of IgA nephropathy with ACE inhibitors: A randomized and controlled trial. J Am Soc Nephrol. 2003; 14: 1578–1583.
- Trimarchi H. Abatacept in glomerular diseases. The open road for the second signal as a new target is settled down. Recent Patents on Endocrine, Metabolic & Immune Drug Discovery. 2015; 9: 1-13.
- Morath C, Zeier M. When should post-transplantation proteinuria be attributed to the renal allograft rather than to the native kidney? Nat Clin Pract Nephrol. 2007; 3: 18–19.
- Reichel H, Zeier M, Ritz E. Proteinuria after renal transplantation: Pathogenesis and management. Nephrol Dial Transplant. 2004; 19: 301–305.
- Fresnedo GF, Escallada R, Rodrigo E, De Francisco AL, Cotorruelo JG. The risk of cardiovascular disease associated with proteinuria in renal transplant patients. Transplantation. 2002; 73: 1345–1348.
- Halimi JM, Matthias B, Al-Najjar A, Laouad I, Chatelet V, Marlière JF, et al. Respective predictive role of urinary albumin excretion and no albumin proteinuria on graft loss and death in renal transplant recipients. Am J Transplant. 2007; 7: 2775–2781.
- KDIGO. Clinical Practice Guideline for the Care of Kidney Transplant Recipients. Am J Transplant. 2009; 9: 1-157.