
Review Article
Austin J Nephrol Hypertens. 2016; 3(1): 1056.
(Pro)renin Receptor and Oxidative Stress Friend or Foe?
Murakami K*
Department of Health Care and Preventive Medicine, Matsuyama Red Cross Hospital, Japan
*Corresponding author: Kazuo Murakami, Department of Health Care and Preventive Medicine, Matsuyama Red Cross Hospital, 1 Bunkyo-cho, Matsuyama, Ehime, 790-8524, Japan
Received: June 19, 2016; Accepted: July 25, 2016; Published: July 27, 2016
Abstract
The renin-angiotensin-aldosterone system (RAAS) plays pivotal role in the pathogenesis of hypertension and renal disease. Oxidative stress is one of the important mechanisms of renal diseases induced by activated RAAS. Recently (Pro)renin receptor (PRR) has been identified, and its importance in the initiation and progression of renal diseases is attracting attentions. Although PRR causes renal injury by increased oxidative stress through Ang II-dependent and independent mechanism, genetic defect in PRR is reported to causes abnormal phenotypes. Recently, PRR has been reported to activate vacuolar H+- ATPase (V-ATPase), that is essential for survive of cells as proton transporter, contributing to keeping cellular pH homeostasis. Loss of V-ATPase activity has been also reported to result in increased oxidative stress. Thus activating PRR may suppress oxidating stress through V-ATPase activation. Actually, loss of this V-ATPase activity has been reported to result in defects in CNS, renal tubular acidosis, osteoporosis, and others. We would also like to show the evolutional relationship between ATP synthase of mitochondria and V-ATPase. ATP synthase of mitochondria resembles V-ATPase in construction but direction of proton flow is opposite. And only V-ATPase has ATP6ap2 as associate protein from PRR and anti-oxidative property. This strange resemblance in structure and opposite anti-oxidative capacity and proton flow across cell membrane reminds us the possibility that mitochondria originally had V-ATPase in cell membrane, but lost ATP6ap2 and proton flow reversed, resulting in loss of antioxidative capacity and gaining of ATP producing capacity in the process of symbiosis and retrogression of mitochondrial DNA.
Keywords: Renin-angiotensin-aldosterone system; Oxidative stress; Reactive oxygen species (ROS); (Pro)renin receptor (PRR); V-ATPase; ATP6ap2
Introduction
Hypertension is a common but one of the most important health problems, because it is a major risk factor for cardiovascular diseases (CVDs) and renal diseases. The renin-angiotensin-aldosterone system (RAAS) plays an important role in the initiation and progression of hypertension and target organ damage [1], although RAAS plays a critical role in controlling blood pressure or hydro-electrolyte balance. And RAAS, not only in the systemic circulation but also in the local organs and tissues, plays a crucial role in the pathogenesis of hypertension, CVDs, and renal diseases [2-4].
Particularly, production of reactive oxygen species (ROS) such as superoxide anion by increased angiotensin II (Ang II) of the classical arm of RAAS is one of the important mechanisms in the pathogenesis of CVDs and renal diseases [5]. And in the past decade, (Pro)renin receptor (PRR) has been identified and its importance in the renal pathophysiology caused by hypertension or diabetes mellitus has been reported. Although PRR causes renal injury partially by increased oxidative stress by Ang II -dependent and independent activations of local RAAS, genetic defect in PRR causes nerval or occular abnormality and even results in fatal. Recently, PRR has been also reported to activate V-ATPase that is essential for survive of cells as proton transporter across cell or organelle membrane resulting in extracellular and organelle acidification, and constituting cellular pH homeostasis [6]. Moreover, loss of this V-ATPase is reported to result in increased oxidative stress in addition to impaired cellular pH homeostasis [7,8]. Thus PRR may suppress excessive oxidating stress through V-ATPase.
We will discuss the mechanism and clinical relevance of these contradictory effects of these PRR on oxidative stress from the viewpoint of recent findings such as PRR, V-ATPase, oxidative stress, acidification mechanism by H+ transporter.
Biology of (pro)renin receptor and oxidative stress
Receptor protein for renin and prorenin, PRR, causing biological effect of renin other than classical arm of RAAS in Ang II -dependent and independent ways, was identified from human kidney in 2002. PRR is a 350-amino acid single transmembrane receptor protein, expressed in brain, heart, lung, liver, kidney, skeletal muscle, pancreas, fat, and placenta. Both prorenin and renin binds to the PRR [9]. After binding to PRR, nonproteolytic activation and conformational change of prorenin occur without cleavage of the prosegment, causing local Ang II generation and Ang II -dependent activation of tissue RAAS [10]. This may lead to increase oxidative stress through activation of AT1 receptor. After the binding of prorenin and renin to PRR as ligands, Ang II -independent signaling cascades are activated. Ang II-independent MAPK activation by human PRR and induction of glomerulosclerosis with increased TGF-beta1 expression was reported [11]. And renin-activated induction of ERK1/2 through a receptor-mediated, angiotensin II-independent mechanism in mesangial cells has been reported. This renin-activated pathway was reported to have triggered cell proliferation along with TGF-beta1 and plasminogen activator inhibitor-1 gene expression [12]. These Ang II -independent signaling pathways may also cause oxidative stress and further enhance end organ damage. Ichihara, et al. reported that the binding of renin and prorenin to the PRR in diabetic nephropathy were inhibited by a decoy peptide corresponding to the “handle” region (HRP) for nonproteolytic activation of prorenin on PRR, and non-proteolytic activation of prorenin may be a significant mechanism of diabetic nephropathy and may serve as important therapeutic targets for the prevention of diabetic organ damage [13]. PRR may affect on vacuolar H+ -ATPase (V-ATPase) which regulates the pH of cell and intracellular organelle [6], because hydrophobic membrane-binding fragment of PRR degraded by furin contains ATPase associated protein 2 (ATP6ap2). Bafilomycin, a specific inhibitor of V-ATPase, has been reported to inhibit phosphorylation of ERK by prorenin in the kidney [14]. Prorenin and its receptormediated Ang II-independent pathways comprise of PRR-associated V-ATPase-linked Wnt/Frizzled signal transduction, including canonical-β-catenin and non-canonical Wnt-JNK-Ca++ signals in the pathogenesis of cardiovascular and renal end-organ damage [15].
Although PRR plays a harmful role in the pathogenesis of renal diseases such as diabetic nephropathy, mutant of PRR is reported to have various abnormal phenotype. So it is suspected that PRR has some important function for cells to survive independent of RAAS. For example, abnormal pigmentation of skin or eye, neural cell death in zebrafish [16], malformation of head and tail, abnormal pigmentation of skin or eye in xenopus laevis [17], X linked recessive familial epilepsy in human [18,19], fulminant heart failure in mouse [20], have been reported. Since mutant of V-ATPase subunit in zebrafish shows similar phenotype as PRR mutant of zebrafish [17], V-ATPase seems have associated in phenotype of PRR mutant. Defect in acidification of organelle and others may be involved for that abnormal phenotype in PRR mutant. Mutations in the gene encoding subunit of V-ATPase are also reported to cause renal tubular acidosis with sensorineural deafness [21], infantile malignant osteopetrosis [22], and osteoporosis [23]. Interestingly, already in 1995 it was reported that inhibitor of V-ATPase, baflomycin, proteolytically processed mutant β-amyloid from familial Alzheimer’s disease differently from wild-type one, both transfected to kidney cells [24]. And X-linked Parkinsonism caused by altered splicing of ATP6ap2 has been also reported [25].
But some reports show that PRR is regulating the production of intracellular ROS such as superoxide anion. In Yeast, mutants lacking V-ATPase subunits results in increased oxidative stress (may be extra-mitochondrial origin) [26,27]. Possible mechanism may be because positively charged cell membrane attracts intracellular electron (from NADH, FADH2, electron donors) to the cytosolic face of plasma membrane electrically due increased H+ concentration of exoplasmic face of the plasma membrane as V-ATPase associatedprotein from PRR activates V-ATPase and facilitate outward flow of H+. Thus decreased intracellular electron cause reduction in generation of intracellular ROS such as superoxide anion from triplet oxygen molecule, because intracellular triplet oxygen molecules interact less frequently with electron donors (Figure 1).
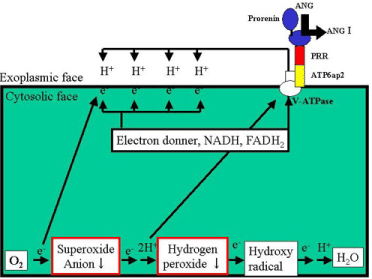
Figure 1: Mechanism of increased antioxidative stress by activating PRR and
V-ATPase. (Author’s speculation).
PRR: (Pro)renin receptor. ATP 6ap2: ATPase associated protein 2.
V-ATPase: vacuolar H+-ATPase.
The binding of renin or prorenin to the PRR is reported to ROS formation through both Ang II -dependent and independent mechanisms. Authors of this report showed that PRR-mediated Ang II -independent ROS formation is associated with activation of the MAPK/ERK1/2 and PI3/Akt signaling pathways using neuronal cells [28].
Is effect of V-ATP independent with PRR?
It has been reported that full-length PRR is cleaved in the trans- Golgi by furin intracellularly into a soluble form of 28 kDa PRR (sPRR) and protein-binding hydrophobic domain [29]. Already before PRR was identified in 2002, 8.9 kDa protein was reported to bind to V-ATPase and the gene coding this protein was named as ATP6ap2 [30]. Truncated hydrophobic protein generated by cleavage of full-length PRR by furin turned out to contain ATP6ap2. So it is suggested full-length PRR is cleaved into sPRR that can activate prorenin non-enzymatically on the cell membrane and ATP6ap2 that binds to and activates V-ATPase. Recently Trepiccione, et al. reported that nephron specific deletion of ATP6ap2 caused decreased V-ATPase expression and activity, down regulation of the medullary NKCC2, autophagic defects, renal tubular acidosis using epithelial cells of medullary tubules of mouse. And interestingly, this nephron specific deletion of ATP6ap2 did not affect Ang II production, Ang II -dependent Blood Pressure regulation, or sodium handling in the kidney [31]. These findings indicate that suppression of V-ATPase causes various renal abnormalities even with unchanged RAAS activity, and malfunction of V-ATPase itself does not affect RAAS activity.
Clinical relevance
Although reports on clinical impact of pH regulation and blood pressure other than RAAS are not seen so far, I would like to show our data with regard to this issue. Figure 2 shows association between urinary pH (pH), systolic blood pressure (SBP), and serum potassium concentration (k) in 12,714 patients who visited our hospital for the first time for the purpose of medical checkup (Figure 2A,B) [32]. PH and SBP correlated positively, SBP and K correlated positively, and pH and K correlated negatively (statistically significant for all). These correlations cannot be explained by RAAS only, because PH and SBP should correlate negatively, SBP and K should correlated negatively, and pH and K should correlate positively to explain this result by the change of renin, angiotensin, and aldosterone. So there must be some confounding factor other than RAAS in these relationships of these factors. V-ATPase may be one possible mechanism for this result, because impairing of it may cause impaired urine acidification and increased oxidative stress leading to elevated SBP, independent of RAAS.
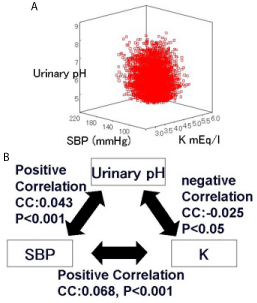
Figure 2: Correlation between urinary pH, systolic blood pressure, and K.
CC: correlation coefficient.
Evolutional consideration for V-ATPase and mitochondrial ATP synthase
There is a strange resemblance between V-ATPase and mitochondrial ATP synthase. Both proton transporters have similar structure and polarity but have reverse flow direction of H+ across the cell membrane. And also V-ATPase consumes ATP and has anti-oxidative stress property, but ATP synthase generates ATP and produces ROS (Figure 3). There is less possibility that these resemblance in structure with opposite flow direction of p H+ and function including ATP and ROS generation have occurred only by chance. Originally, these two kinds of transporters may have been the same one, V-ATPase on the cell membrane of differently living cells, ancestor of mitochondria and eukaryote. Both V-ATPase may have had same important function as anti-oxidant consuming ATP. But after ancestor of mitochondria has been incorporated into cytosol of eukaryote and began symbiosis with eukaryote, mitochondrial V-ATPase may have lost its ATP6ap2/PRR in the process of retrogression of mitochondrial DNA, and may not be able to keep outward driving force of H+ any longer. And reverse flow of proton may have occurred due to concentration gradient of H+ between inter membrane space and matrix. Then reverse rotation of V-ATPase may have occurred, turning motor into generator. Thus mitochondrial ATP synthase may have obtained ATP-generating capacity, but lost anti-oxidative capacity in exchange. This incident may have benefitted both eukaryote host and mitochondria to survive.
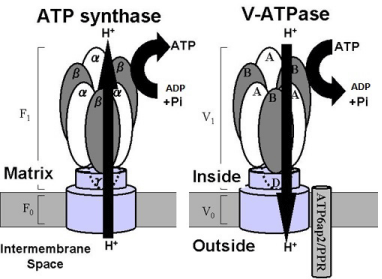
Figure 3: Overall survival, autologous stem cell transplant (ASCT) versus no ASCT (p=0.12).
Balance and imbalance of pro- and anti-oxidative effects of PRR in normal and diseased state
Keeping biological homeostasis by balance of pro- and antioxidative mechanism is important issue. Unfortunately, at present, no attention is paid about the clinical importance of anti-oxidative role of V-ATPase and there is no clinical report demonstrating directly the protective role from oxidative stress through V-ATPase activation by PRR, although A II -dependent and A II -independent increase of oxidative stress by PRR has been reported. So it is hard to directly identify oxidative stress change by V-ATPase as a cause of disease clinically at present, although protective role of V-ATPase activation by anti-oxidative mechanism has been reported using yeast experimentally [26,27]. As many reports have demonstrated that animal model with genetic mutation of PRR and V-ATPase has shown similar abnormal phenotypes, common pathway through PRR and V-ATPase leading to these phenotypes is suspected. But it is not fully investigated as to whether this pathway includes oxidative stress or disturbance of pH homeostasis at present. Interestingly, although efficacy of HRP has been reported in diabetic model rat [12], HRP has been reported to have failed in protecting target organ damage in transgenic rat of human renin and angiotensinogen [33]. This may suggest that pro-oxidative stress or other mechanism through PRR may be augmented A II -dependently and A II -independently, A II -dependent and A II -independent pro-oxidative effects by PRR and anti-oxidative effects by V-ATPase may be balanced and keeping homeostasis in non-diabetic or normal state. But possibility of total low contribution of both PRR and V-ATPase to target organ damage in non-diabetic state cannot be ruled out. On top of that, failure of target organ damage protection is also reported in renovascular hypertension model animal model [34]. In addition to abovementioned mechanism, active renin may be endogenous inhibitor in generating more active renin from prorenin and A II at PRR in local RAAS and this may be another fail-safe system of keeping homeostasis of oxidative stress in systemic and circulating high renin state.
Conclusion
PRR generates ROS through Ang II -dependent and Ang II -independent pathway. But PRR itself can also decrease oxidative stress as an associated protein for V-ATPase, activating it. Activating V-ATPase can lead to decreasing oxidative stress.
Strangely enough, V-ATPase and mitochondrial ATP synthase have structural resemblance in common, in addition to opposite function concerning ATP and anti-oxidative property. This may tell something about evolution of mitochondrial ATP synthase.
References
- Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006; 86: 747-803.
- Carey RM. The intra renal renin-angiotensin system in hypertension. Adv Chronic Kidney Dis. 2015; 22: 204-210.
- Aroor AR, Demarco VG, Jia G, Sun Z, Nistala R, Meininger GA, et al. The role of tissue Renin-Angiotensin-aldosterone system in the development of endothelial dysfunction and arterial stiffness. Front Endocrinol (Lausanne). 2013; 4: 161.
- Endo-Mochizuki Y, Mochizuki N, Sawa H, Takada A, Okamoto H, Kawaguchi H, et al. Expression of renin and angiotensin-converting enzyme in human hearts. Heart Vessels. 1995; 10: 285-293.
- Murakami K. New Components of the Renin-Angiotensin- Aldosterone System and Oxidative Stress. J Hypertens (Los Angel). 2015: 4: 211.
- Ichihara A, Sakoda M, Kurauchi-Mito A, Narita T, Kinouchi K. Possible roles of human (pro)renin receptor suggested by recent clinical and experimental findings. Hypertens Res. 2010; 33: 177-180.
- Kane PM. The long physiological reach of the yeast vacuolar H+-ATPase. J Bioenerg Biomembr. 2007; 39: 415-421.
- Milgrom E, Diab H, Middleton F, Kane PM. Loss of vacuolar proton-translocating ATPase activity in yeast results in chronic oxidative stress. J Biol Chem. 2007; 282: 7125-7136.
- Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002; 109: 1417-1427.
- Saris JJ, van den Eijnden MM, Lamers JM, Saxena PR, Schalekamp MA, Danser AH. Prorenin-induced myocyte proliferation: no role for intracellular angiotensin II. Hypertension. 2002; 39: 573-577.
- Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN, et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol. 2007; 18:1789-1795.
- Huang Y, Noble NA, Zhang J, Xu C, Border WA. Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int. 2007; 72: 45-52.
- Ichihara A, Hayashi M, Kaneshiro Y, Suzuki F, Nakagawa T, Tada Y, et al. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the "handle" region for nonproteolytic activation of prorenin. J Clin Invest. 2004; 114: 1128-1135.
- Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, et al. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension. 2009; 54: 261-269.
- Jagadeesh G, Balakumar P, Stockbridge N. How well do aliskiren's purported mechanisms track its effects on cardiovascular and renal disorders? Cell Signal. 2012; 24: 1583-1591.
- Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004; 101: 2792-1279.
- Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, et al. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010; 327: 459-463.
- Contrepas A, Walker J, Koulakoff A, Franek KJ, Qadri F, Giaume C, et al. A role of the (pro)renin receptor in neuronal cell differentiation. Am J Physiol Regul Integr Comp Physiol. 2009; 297: R250-257.
- Ramser J, Abidi FE, Burckle CA, Lenski C, Toriello H, Wen G, et al. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum Mol Genet. 2005; 14: 1019-1027.
- Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Kurauchi-Mito A, et al. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res. 2010; 107: 30-34.
- Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999; 21: 84-90.
- Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, et al. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000; 9: 2059-2063.
- Ochotny N, Van Vliet A, Chan N, Yao Y, Morel M, Kartner N, et al. Effects of human a3 and a4 mutations that result in osteopetrosis and distal renal tubular acidosis on yeast V-ATPase expression and activity. J Biol Chem. 2006; 281: 26102-26111.
- Haass C, Capell A, Citron M, Teplow DB, Selkoe DJ. The vacuolar H(+)-ATPase inhibitor bafilomycin A1 differentially affects proteolytic processing of mutant and wild-type beta-amyloid precursor protein. J Biol Chem. 1995; 270: 6186-6192.
- Korvatska O, Strand NS, Berndt JD, Strovas T, Chen DH, Leverenz JB, et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum Mol Genet. 2013; 22: 3259-3268.
- Kane PM. The long physiological reach of the yeast vacuolar H+-ATPase. J Bioenerg Biomembr. 2007; 39: 415-421.
- Milgrom E, Diab H, Middleton F, Kane PM. Loss of vacuolar proton-translocating ATPase activity in yeast results in chronic oxidative stress. J Biol Chem. 2007; 282: 7125-7136.
- Peng H, Li W, Seth DM, Nair AR, Francis J, Feng Y. (Pro)renin receptor mediates both angiotensin II-dependent and -independent oxidative stress in neuronal cells. PLoS One. 2013; 8: e58339.
- Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension. 2009; 53:1077-1082.
- Ludwig J, Kerscher S, Brandt U, Pfeiffer K, Getlawi F, Apps DK, et al. Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J Biol Chem. 1998; 273: 10939-10947.
- Trepiccione F, Gerber SD, Grahammer F, López-Cayuqueo KI, Baudrie V, Păunescu TG, et al. Renal Atp6ap2/(Pro)renin Receptor Is Required for Normal Vacuolar H+-ATPase Function but Not for the Renin-Angiotensin System. J Am Soc Nephrol. 2016.
- Murakami K. The 39th Annual Scientific Meeting of the Japanese Society of Hypertension, October 2nd, 2016; Japan.
- Feldt S, Maschke U, Dechend R, Luft FC, Muller DN. The putative (pro)renin receptor blocker HRP fails to prevent (pro)renin signaling. J Am Soc Nephrol. 2008; 19: 743-748.
- Muller DN, Klanke B, Feldt S, Cordasic N, Hartner A, Schmieder RE, et al. (Pro)renin receptor peptide inhibitor "handle-region" peptide does not affect hypertensive nephrosclerosis in Goldblatt rats. Hypertension. 2008; 51: 676-681.