
Mini Review
Austin J Nephrol Hypertens. 2016; 3(2): 1059.
Necroptosis in Acute Kidney Injury
Liu W¹* and Xia Y²*
¹School of Medicine, Yunnan University, China
²School of Biomedical Sciences, Chinese University of Hong Kong, China
*Corresponding author: Wenjing Liu, School of Medicine, Yunnan University, Kunming, Yunnan 650091, China
Yin Xia, School of Biomedical Sciences, Chinese University of Hong Kong, Shatin, N.T., Hong Kong, China
Received: August 02, 2016; Accepted: September 05, 2016; Published: September 07, 2016
Abstract
Acute kidney injury (AKI) is a common clinical entity associated with high morbidity and mortality. Tubular epithelial cell death plays a key role in the development of AKI. Although apoptosis has been the focus of the studies on kidney injury and drug discovery for many years, recent studies have identified signaling pathways and regulatory mechanisms for programmed necrosis or necroptosis. It is now clear that receptor-interacting kinase 1 (RIP1), RIP3 and its substrate, the pseudokinase mixed lineage kinase domain-like protein (MLKL) are the core components of necroptosis. Increasing evidence from pharmacological and genetic studies shows that necroptosis plays critical roles in progression of AKI in several mouse models. In this review, we aim to briefly summarize the mechanisms and functions of necroptosis in AKI.
Keywords: Acute kidney injury; Necroptosis; Loss and End-stage kidney disease
Introduction
Acute kidney injury
Acute kidney injury (AKI) is characterized by rapid declines in kidney functions within one week. AKI affects millions of patients worldwide with high mortality, morbidity and cost. Stages of kidney failure in AKI are defined using either Risk, Injury, Failure, Loss and End-stage kidney disease (RIFLE) staging criteria or Acute Kidney Injury Network (AKIN) staging criteria. The RIFLE acronym represents the rising severity grades: risk, injury and failure, and the two outcome criteria: loss and end-stage kidney disease. The severity grades from risk to failure are defined based on increasing serum creatinine and decreasing urine output. The two outcome categories, loss and end-stage kidney disease, are defined by the duration of loss of kidney function [1]. However, the AKIN staging system includes the entire spectrum of symptoms, from tiny impairment in kidney function to the need for renal replacement therapy [2].
Using the RIFLE classification, studies from all around the world revealed that 2-7% of the hospitalized patients suffered from AKI, and the incidence is steadily increasing [3]. From the studies of the incidence of AKI in an intensive care unit in Australia from 2000 to 2005, 36.1% of patients suffered from AKI [4]. AKI is associated with an increased mortality in hospitalized patients.
The cause of acute kidney injury is grouped into three classes: prerenal, renal (with direct intrinsic renal damage) and postrenal injury [5]. Prerenal acute kidney injury is caused by diseases in cardiovascular system such as systemic hypotension, severe systolic cardiac failure and volume depletion, which lead to reduction in renal perfusion and glomerular filtration. Intrinsic acute kidney injury is the consequence of destruction of nephron structure, and it results from ischemic or nephrotoxic injury, such as acute progressive glomerulopathies, acute interstitial nephritis and acute tubular necrosis. Postrenal acute kidney injury is attributed to obstruction of the urinary tract, which causes reduced glomerular filtration rate and renal failure.
Approximately one third of the AKI result from direct or indirect nephrotoxicity, and two-thirds result from renal ischemiareperfusion or sepsis [6]. Although AKI has various etiologies, it is frequently associated with ischemic and toxic insults. AKI occurs commonly in the setting of sepsis. The pathophysiology of sepsisassociated AKI is very complex, but it involves the changes as seen in ischemic, toxic or obstructive nephropathy, including inflammatory response, oxidative stress and microvascular dysfunction [7]. In laboratory science, the molecular events of AKI are most commonly studied in rodents with ischemia-reperfusion injury induced by clamping of both renal pedicles. Other models used in laboratories include cisplatin- or folic acid-induced toxic injury and sodium oxalate-induced crystal nephropathy. Accordingly, our knowledge regarding the mechanisms of AKI has been mostly obtained from studies in rats and mice with ischemia/reperfusion-induced AKI.
Tubular epithelial cells play active roles in the development of AKI. In response to various insults, tubular epithelial cells die, leading to tubular atrophy and even kidney failure [8]. Although acute tubular necrosis is a key feature of AKI, previous studies were directed toward apoptosis because apoptosis was considered to be the only genetically programmed and therapeutically targetable form of cell death in AKI while necrosis was thought to be a nonregulated response to overwhelming stress. Apoptosis is a caspase-dependent programmed cell death, and is characterized by cell shrinkage, chromatin condensation, nuclear fragmentation and membrane blebbing [9]. Apoptosis of renal IRI was first described in 1992 [10], and subsequently it was shown that blockade of apoptosis with IGF-1 or the pan caspase inhibitor zVAD-fmk prevented renal function impairment after IR [11]. Therefore, for a long time, strategies targeting the apoptosis pathway were widely explored for AKI treatment. Despite the substantial therapeutic effect in animal models, efficient anti-apoptosis intervention strategies are still absent in clinic.
Studies in the past decade have found that necrosis can be a regulated or programmed process. Receptor-interacting protein kinase (RIP)1 and RIP3-mediated necroptosis [12,13] is the best studied regulated necrosis pathway. Necroptosis is characterized by cytoplasmic granulation and organelle or cellular swelling that together result in cell membrane rupture and release of intracellular contents [14]. Unlike apoptotic cells, which are rapidly phagocytosed by macrophages or neighboring cells, necrotic cell death including necroptosis provokes sterile inflammation by the damage-associated molecular patterns (DAMPs) formulated from the released intracellular contents. Therefore, necroptosis not only participates in the development of organism but also is critically involved in various pathological processes including ischemic injury in brain, heart and kidney [12,15-18], atherosclerosis [19], pancreatitis [20], inflammatory bowel diseases [21] and viral infection [22]. Emerging evidence indicates that AKI involves necroptosis, and that the necroptotic pathway may be used for therapeutic intervention to limit AKI [17,18,23,24].
The pathways of necroptosis
As shown by Figure 1, necroptosis can be activated when cells are treated with TNF family cytokines, including TNFα, Fas ligand and TNF-related apoptosis-inducing ligand (TRAIL) [25]. Ligation of TNF to TNF receptor (TNFR)1 causes formation of receptorassociated complex I, which contains TNFR1, TNFR1-associated death domain (TRADD), TNFR-associated factor 2 (TRAF2), cellular inhibitor of apoptosis protein (cIAP) 1, cIAP2, the linear ubiquitin chain assembly complex (LUBAC) and RIP1. CIAPs and LUBAC promote RIP1 ubiquitination at Lys-11 and Lys-63, and linear ubiquitin linkages. Ubiquitination of RIP1 further recruits additional factors, including transforming growth factor (TGF) β-activated kinase (TAK1), TAK1-binding protein 2 (TAB2) and the inhibitor of the NF-κB kinase (IKK), and ultimately activates the NF-κB pathway [26].
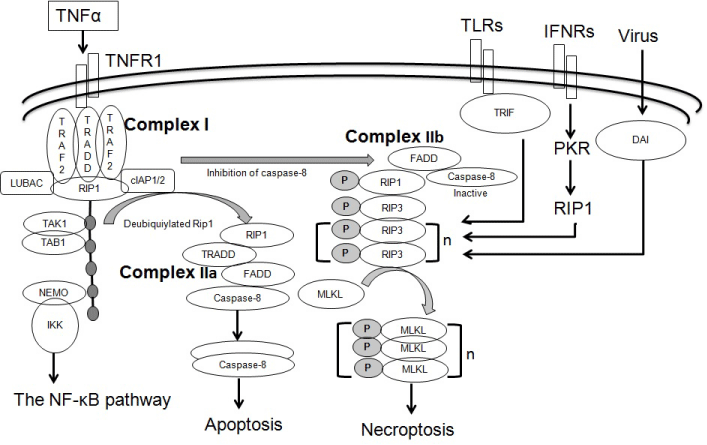
Figure 1: The pathways of necroptosis. Binding of TNFα to its receptor TNFR1 triggers the assembly of Complex I, which comprises TNFR1, TRADD, TRAF2,
cIAP1/2, LUBAC and RIP1. Ubquitylated RIP1recruits TAK1, TAB1, IKK and NEMO, thus activating the NF-?B pathway. When RIP1 is deubiquitylated, Complex
IIa is formed. Complex IIa consists of TRADD, FADD, RIP1 and caspase-8, and activates the caspase-8 and apoptosis pathway. When the caspase-8 activity is
inhibited, Complex IIb is formed. Complex IIb entails the association of RIP1 and RIP3 followed by series of phosphorylation events. Activated RIP3 recruits and
phosphorylates MLKL. Phosphorylation of MLKL induces its oligomerization, membrane association and neceroptosis.TLR3, TLR4, IFNRs and the intracellular
RHIM-containing protein DAI also induce necroptosis. Diverse upstream stimuli converge on the RIP3-MLKL pathway.
Alternatively, complex I may lead to cell death signaling. When the polyubiquitin chain on RIP1 is removed by cylindromatosis (CYLD) or the ubiquitin-modifying enzyme A20, RIP1 binds to the FAS-associated death domain (FADD) to form a cytosolic deathinducing signaling complex (DISC, as known as complex IIa), which consists of TRADD, FADD, caspase-8 and RIP1 [27]. The formation of complex IIa leads to activation of caspase-8 and apoptosis.
When the caspase-8 activity is inhibited under some pathophysiological conditions, complex IIb (also known as necroptosome or necrosome) forms to activate the necroptotic pathway [28-30]. In this case, RIP1 and RIP3 bind each other through their respective homotypic interaction motif (RHIM) domain and through a series of RIP1 and RIP3 auto-and trans-phosphorylation events. Phosphorylation of RIP3 at Thr-231/Ser-232 (Ser-232 in human RIP3) [31-34] stimulates the recruitment of the downstream substrate mixed lineage kinase-domain like (MLKL). MLKL is subsequently phosphorylated at Thr 357 and Ser 358 in humans [31] or at Ser 345 and Ser 347 in mice [34]. Phosphorylation of MLKL triggers its oligomerization and membrane association, which are necessary and sufficient for the induction of necrosis [20,23,31,35- 39]. In most pathological conditions, TNFR signals are relayed to apoptosis rather than necroptosis. Only when apoptosis is blocked, TNFR signals may lead to necroptosis.
Several different pathways can also induce RIP3-dependent necroptosis through distinct mechanisms. For instance, Toll-like receptor (TLR) 3 or TLR4 directly activates RIP3 kinase through a RIP homotypic interaction motif known as TRIF, and this pathway was found to be independent of RIP1 or its kinase activity in fibroblasts [38,40]. Interferons (INFs) induce RIP1/RIP3–mediated necrosis through the RNA-responsive protein kinase PKR. It has been reported that IFN-induced necrosis proceeds via progressive assembly of a RIP1–RIP3 “necrosome” complex that requires Jak1/ STAT1-dependent transcription, but does not need the kinase activity of RIP1. Instead, IFNs transcriptionally activate the RNAresponsive protein kinase PKR, which then interacts with RIP1 to initiate necrosome formation and trigger necrosis [41,42]. The RHIM-containing cellular protein DNA-dependent activator of interferon regulatory factors (DAI, also known as ZBP1 or DLM- 1) can also interact with RIP3 directly, following infection with murine cytomegalovirus (MCMV) [22,43]. It is clear the RIP3 kinase activation plays an important role in the initiation of that necrosis. Activated RIP3 then transduces the necrosis signal to MLKL.
Virally infected cells are mostly cleared through extrinsic apoptosis, which involves caspase-8 activation and the downstream effectors caspase-3, caspase-6 and caspase-7. However, some viruses including vaccinia virus, cytomegalovirus, cowpox virus, express inhibitors of caspases. In cells infected with these virus, apoptosis is no longer induced, and spontaneous necroptosis is initiated to function as a second-line defense mechanism against caspase-inhibitorexpressing viruses [43-45]. Therefore, the necroptosis pathway acts as an alternative “fail-safe” cell death pathway in cases where cells are unable to undergo apoptosis, when the apoptosis signaling is blocked by endogenous or exogenous factors such as viruses or mutations.
MLKL consists of an N-terminal 4-helical bundle domain (4HBD) fused by a brace region to a C-terminal pseudokinase domain. The 4HBD of MLKL is structurally similar to a-pore-forming toxins, and is sufficient to oligomerize, bind to phosphatidylinositol lipids through a patch of positively charged amino acids, permeabilize the plasma membrane and trigger cell death [35-37,46,47]. Once MLKL is phosphorylated on its C-terminus, 4HBD is unleashed from the closed conformation of MLKL to target the plasma membrane and subsequent membrane rupture [46]. There are two non-exclusive models to explain the consequences of the association of MLKL and the plasma membrane. First, MLKL oligomers act directly as a poreforming complex, contributing to the membrane destabilization. Second, oligomerized MLKL deregulates Ca2+ or Na+ ion channels, resulting in ion influx and eventual plasma membrane disruption [36,37].
The regulators of necroptosis
Inhibitors and activators of necroptosis can be clinically used for a wide range of necroptosis-related diseases. Therefore, intensive efforts for the discovery of small molecules with potency and specificity have been made over the last decade or so. Degterev, et al. [12] identified for the first time the compound necrostatin-1 (Nec-1) that inhibits RIP1 and necroptosis by screening 15,000 compounds. However, Nec-1 is relatively toxic and not very specific for necroptosis. Nec- 1s is a derivative of Nec-1, and has increased specificity for RIP1 over a broad range of kinases, thus, Nec-1s is a preferred tool for targeting RIP1 in vivo [48]. A cell-permeable acrylamide compound necrosulfonamide (NSA) selectively binds to and inhibit human (not mouse) MLKL function via covalent modification of Cys86 [31]. More recently, the ability of the first-identified series of RIP3 inhibitors was tested, and this led to the identification of compounds GSK’840, GSK’843 and GSK’872 [49]. However, GSK’840 is unable to inhibit murine RIP3.
Besides direct necroptosis inhibitors, other approaches that interfere with the necroptotic pathway have also been sought. It has been reported that Rhenium (V) oxo complexes of general formula [ReO(OMe)(N^N)Cl2] effectively killed cancer cells by triggering necroptosis. The complexes evoke necrosome-dependent intracellular reactive oxygen species (ROS) production and propidium iodide uptake [50]. ROS are critical regulators of necroptotic signaling. ROS scavengers such as butylated hydroxyanisole, N-acetylcysteine, α-tocopherol and ethyl pyruvate significantly inhibited TNFα-induced necroptotic signaling and cell death [51]. Autophagy may serve as a pro-survival mechanism, delaying the induction of necroptosis in melanoma cells. The plant quaternary benzo [c] phenanthridine alkaloid sanguilutine (SL) was found to be a strong inducer of necroptotic death in human melanoma cells. In addition, SL triggered an autophagic response. Interestingly, combined treatment with SL and the autophagy inhibitors 3-methyladenine, bafilomycin-A1 and LY294002 increased necroptotic cell death [52]. Shikonin, a naturally occurring compound, induced cell death through the necroptotic pathway in MCF-7 and HEK293 [53].
Necroptosis in AKI
Necroptosis has been found in a number of animal models of acute kidney injury including ischemia/reperfusion injury (IRI) [17,18], cisplatin nephrotoxicity [18,23] or crystal-induced cytotoxicity [24]. RIP1 and RIP3 protein expression in the proximal tubules was found to be induced by IR [17,18]. Inhibition of necroptosis by injection of Nec-1either before or after ischemia protects from AKI, as demonstrated by reduced tubular damage and renal functional impairment, and prolonged survival [17]. Knockout of the key necroptotic gene Rip3 in mice also protected against IRI [18]. In vitro, TNF-α, cycloheximide and zVAD (TCZ) induced necroptosis in TKPTS mouse proximal tubular cells [17]. Nec-1 inhibited cell death induced by TNF-α-stimulation and ATPdepletion in rat tubular NRK-52 cells and human tubular epithelial HK-2 cells [54,55]. Hypoxia/reoxygeneation or TNF-α, TWEAK and INF-γ (TTI) also caused necroptotic death in freshly isolated renal tubules [17,22]. Therefore, it is clear that IRI involves tubular cell necroptosis. Interestingly, blockade of apoptosis by zVAD, a pancaspase inhibitor, did not alleviate the renal dysfunction and tissue damage [17]. Furthermore, caspase-8/Rip3-double knockout mice did not provide additional protection over Rip3 single knockout in the ischemic model. In addition, no cleaved caspase-3 was found in the time course of IRI [17]. These results suggest that necroptosis plays an important role in IRI, while apoptosis may be of minor importance in IRI [18].
A previous study by Daemen and colleagues found that zVAD prevented renal apoptosis, inflammation and tissue injury in IRI [11]. The discrepancy between the two studies using zVAD remains unexplained. One possible reason is the different methods to induce IRI in these studies, which may result in different types of cellular death in kidneys [56].
The presence of necroptosis in the toxic kidney injury induced by cisplatin has been demonstrated by several studies. Cisplatin is a widely used chemotherapy drug for solid tumors, but its clinical application is restricted by its nephrotoxicity. Tristao, et al. [57] reported that Nec-1 has no effect on cisplatin-induced apoptosis in HK-2 cells. When apoptosis was blocked by z-VAD, Nec-1 completely reversed cell viability, suggesting that inhibition of apoptosis triggers necroptosis in HK-2 cells. The importance of necroptosis in toxic kidney injury was further demonstrated by a recent study using genetic mouse models [23]. In this study, Xu and colleagues showed that RIP1, RIP3 and MLKL expression in tubular cells and in kidney was induced by cisplatin treatment. Rip3 or Mlkl knockout mice were protected from kidney injury induced by cisplatin. Nec- 1 treatment also prevented cisplatin-induced kidney injury. In vitro, cisplatin directly induced necroptosis in cultured primary proximal tubular cells. Mechanistically, cisplatin induced RIP1 and RIP3 phosphorylation, and promoted the formation of the RIP1, RIP3 and MLKL complex in proximal tubular cells [23].
Mulay, et al. reported that crystals of calcium oxalate, monosodium urate, calcium pyrophosphate dihydrate or cysteine induced RIP1 and RIP3 expression and MLKL phosphorylation, and triggered caspase-independent cell death in mouse kidney tubular cells, which was blocked by Nec-1. In sodium oxalate-induced acute crystal nephropathy (CN), injured epithelial cells exhibited characteristics of necrosis, which was associated with increased expression of RIP1, RIP3 and MLKL. All the functional and structural parameters of acute CN were ameliorated in Rip3 knockout or MLKL knockout mice or by Nec-1 treatment. Interestingly, crystal-induced necroptosis requires TNFR1. These results suggest that the necroptotic TNFR1/ RIP1/RIP3/MLKL pathway may be useful therapeutic targets for CN treatment [24].
Cyclosporin A (CsA) has nephrotoxic effects. CsA is a widely used immunosuppressive drug, and it induces renal tubular epithelial cell death. Ouyang, et al. demonstrated that the majority of NRK- 52E cells died of necrosis in the presence of CsA and treatment with Nec-1 or knockdown of RIP3 significantly reduced the cell death [58]. Therefore, CsA may induce necroptosis in renal tubular cells, but this hypothesis remains to be verified in vivo.
The role of necroptosis has also been investigated in the kidney injury model of glycerol-induced rhabdomyolysis. Homsi, et al. found that the increased serum creatinine levels after injection of glycerol into muscles were attenuated by Nec-1 [59]. However, BUN and histological results were not provided in the study, thus it is unclear whether the attenuated creatinine levels resulted from improved kidney structure and function or from the potential effects of Nec-1 on muscles and even livers.
Perspectives
The necroptosis pathways have been well defined. Studies from cell culture and mouse models clearly demonstrate that AKI involves necroptosis and necroptosis contributes to the pathogenesis of AKI. However, these previous studies used Nec-1 administration, or global Rip3 or Mlkl knockout mouse models, thus the cell-specific sensitivity to necroptosis is unclear. Tubular cells have been shown to undergo necroptosis in vitro by several studies [17,18,23,24], but whether necroptosis is a primary mode of regulated cell death in renal tubules in vivo is questioned by a study showing that conditional deletion of FADD or caspase-8 in renal tubules neither induced spontaneous necroptosis nor sensitized tubular cells to cisplatin-induced cell death [60]. Instead, the protective effects in global Rip3 KO or Nec-1- treated mice were due to increased dilation of peritubular capillaries [60,61]. Nevertheless, whether tubular cells undergo necroptosis in AKI remains to be further investigated using tubular cell specific knockout of Rip3 or Mlkl.
In addition to apoptosis and necroptosis, another type of regulated cell death, i.e., ferroptosis was found to contribute to tubular cell necrosis [60]. Ferroptosis is an iron-dependent form of necrosis that occurs due to lipid peroxidation. Ferroptosis is induced when the glutamate/cysteine antiporter, system xc -, or glutathione peroxidase 4 (GPX4) is inhibited, and is prevented by ferrostatin-1 or iron depletion [62,63]. Therefore, multiple forms of regulated cell death coexist in the pathogenesis of AKI. The dynamics and relative contribution of each form of cell death during the development of AKI remain unknown, but it may depend on the type and severity of the injury. Future work should be directed toward understanding of the profiles and contributions of apoptosis, necroptosis, ferroptosis as well as other types of cell death, so that optimized therapeutic treatments can be eventually developed.
Acknowledgement
This study was supported by an RGC-NSFC joint grant (N_ CUHK432/12), and a CUHK direct grant (4054305).
References
- Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004; 8: R204-212.
- Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007; 11: R31.
- Kerr M, Bedford M, Matthews B, O'Donoghue D. The economic impact of acute kidney injury in England. Nephrol Dial Transplant. 2014; 29: 1362-1368.
- Case J, Khan S, Khalid R, Khan A. Epidemiology of acute kidney injury in the intensive care unit. Crit Care Res Pract. 2013; 2013: 479730.
- Sancho-Martinez SM, Lopez-Novoa JM, Lopez-Hernandez FJ. Pathophysiological role of different tubular epithelial cell death modes in acute kidney injury. Clin Kidney J. 2015; 8: 548-559.
- Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, Kellum JA, et al. Acute kidney injury: an increasing global concern. Lancet. 2013; 382: 170-179.
- Gomez H, Ince C, De Backer D, Pickkers P, Payen D, Hotchkiss J, et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014; 41: 3-11.
- Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011; 121: 4210-4221.
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009; 361: 1570-1583.
- Schumer M, Colombel MC, Sawczuk IS, Gobe G, Connor J, O'Toole KM, et al. Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am J Pathol. 1992; 140: 831-838.
- Daemen MA, van't Veer C, Denecker G, Heemskerk VH, Wolfs TG, Clauss M, et al. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest. 1999; 104: 541-549.
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005; 1:112-119.
- Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. 2014; 25: 2689-2701.
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010; 11: 700-714.
- Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007; 21: 227-233.
- Kung G, Konstantinidis K, Kitsis RN. Programmed necrosis, not apoptosis, in the heart. Circ Res. 2011; 108: 1017-1036.
- Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012; 81: 751-761.
- Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2013; 110: 12024-12029.
- Lin J, Li H, Yang M, Ren J, Huang Z, Han F, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013; 3: 200-210.
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009; 137: 1100-1111.
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011; 477: 330-334.
- Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012; 11: 290-297.
- Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J Am Soc Nephrol. 2015; 26: 2647-2658.
- Mulay SR, Desai J, Kumar SV, Eberhard JN, Thomasova D, Romoli S, et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat Commun. 2016; 7: 10274.
- He S, Huang S, Shen Z. Biomarkers for the detection of necroptosis. Cell Mol Life Sci. 2016; 73: 2177-2181.
- Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell. 2015; 60: 63-76.
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003; 114: 181-190.
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008; 133: 693-703.
- Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013; 20: 1381-1392.
- Legarda-Addison D, Hase H, O'Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ. 2009; 16: 1279-1288.
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012; 148: 213-227.
- Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem. 2013; 288: 16247-16261.
- McQuade T, Cho Y, Chan FK. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem J. 2013; 456: 409-415.
- Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 2013; 5: 70-78.
- Wang X, Li Y, Liu S, Yu X, Li L, Shi C, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A. 2014; 111: 15438-15443.
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014; 16: 55-65.
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014; 24: 105-121.
- He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011; 108: 20054-20059.
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014; 343: 1357-1360.
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013; 288: 31268-31279.
- McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S A. 2014; 111: E3206-3213.
- Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013; 110: E3109-3118.
- Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010; 7: 302-313.
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009; 137: 1112-1123.
- Upton JW, Kaiser WJ, Mocarski ES. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem. 2008; 283: 16966-16970.
- Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014; 111: 15072-15077.
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014; 7: 971-981.
- Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012; 3: e437.
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014; 56: 481-495.
- Suntharalingam K, Awuah SG, Bruno PM, Johnstone TC, Wang F, Lin W, et al. Necroptosis-inducing rhenium(V) oxo complexes. J Am Chem Soc. 2015; 137: 2967-2974.
- Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene. 2015; 34: 5796-5806.
- Hammerova J, Uldrijan S, Taborska E, Vaculova AH, Slaninova I. Necroptosis modulated by autophagy is a predominant form of melanoma cell death induced by sanguilutine. Biol Chem. 2012; 393: 647-658.
- Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007; 6: 1641-1649.
- Zhang L, Jiang F, Chen Y, Luo J, Liu S, Zhang B, et al. Necrostatin-1 attenuates ischemia injury induced cell death in rat tubular cell line NRK-52E through decreased Drp1 expression. Int J Mol Sci. 2013; 14: 24742-24754.
- Liang X, Chen Y, Zhang L, Jiang F, Wang W, Ye Z, et al. Necroptosis, a novel form of caspase-independent cell death, contributes to renal epithelial cell damage in an ATP-depleted renal ischemia model. Mol Med Rep. 2014; 10: 719-724.
- Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011; 80: 29-40.
- Tristao VR, Goncalves PF, Dalboni MA, Batista MC, Durao Mde S, Jr., Monte JC. Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren Fail. 2012; 34: 373-377.
- Ouyang Z, Zhu S, Jin J, Li J, Qiu Y, Huang M, et al. Necroptosis contributes to the cyclosporin A-induced cytotoxicity in NRK-52E cells. Pharmazie. 2012; 67: 725-732.
- Homsi E, Andreazzi DD, Faria JB, Janino P. TNF-alpha-mediated cardiorenal injury after rhabdomyolysis in rats. Am J Physiol Renal Physiol. 2015; 308: F1259-1267.
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014; 111: 16836-16841.
- Linkermann A, Heller JO, Prokai A, Weinberg JM, De Zen F, Himmerkus N, et al. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol. 2013; 24:1545-1557.
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012; 149: 1060-1072.
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014; 156: 317-331.