
Research Article
Austin J Nephrol Hypertens - Volume 8 Issue 1 - 2021
Matrix metalloproteinase 9 Contributes to Glomerulosclerosis by Causing Profibrotic Changes in Podocytes and Glomerular Endothelial Cells
Qiao X1,2#, Guo J1,3#, Chen J1#, Loron MC1,4, Zhao Y1,5, Rao P1, Cao Q1, Wang Y1, Harris DCH1 and Zheng G1*
1Centre for Transplant and Renal Research, The Westmead Institute for Medical Research, the University of Sydney, Sydney, NSW, Australia
2Department of Nephrology, Second Hospital of Shanxi Medical University, Shanxi Kidney Disease Institute, Taiyuan, Shanxi, People’s Republic of China
3Department of Pathophysiology, Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China
4Department of Nephrology, Rouen University Hospital, University of Paris, Paris, France
5The School of Biomedical Sciences, Chengdu Medical College, Chengdu, People’s Republic of China
#Contributed Equally to this Work and are co-First Authors
*Corresponding author: Zheng G, Centre for Transplantation and Renal Research, The Westmead Institute for Medical Research, University of Sydney, Sydney, NSW 2145, Australia
Received: May 18, 2021; Accepted: June 29, 2021; Published: July 06, 2021
Abstract
Background: Glomerulosclerosis is characterized by progressive (myo) fibroblast accumulation and collagen deposition involving profibrotic changes of podocytes and endothelial cells. A profibrotic role of MMP-9 in interstitial fibrosis has been reported. Whether MMP-9 plays a role in glomerulosclerosis is not clear yet.
Methods: Mouse glomerulosclerosis model [Adriamycin Nephropathy (AN) model] was induced by a single adriamycin injection (10.2mg/kg, with physiological saline for controls) through tail vein in MMP-9-/- and wild-type control mice of BALB/c background. All animals were sacrificed at 4 weeks after injection. Albuminuria (albumin to creatinine ratio) and calculated GFR were measured. Gomori Trichrome (GT) and Sirius Red (SR) staining were used for assessment of glomerular fibrosis. Profibrotic changes of podocytes or glomerular endothelial cells were examined by confocal microscopy using immunofluorescence staining (IF) of desmin or a-SMA with P-cadherin or VEcadherin.
Results: Albuminuria was reduced while GFR was increased in MMP-9-/- AN mice compared with those of wild-type mice. Confocal microscopy showed a significant decrease in podocytes double-stained with P-cadherin and desmin, demonstrating that MMP9-/- AN mice were protected from profibrotic changes in podocytes and glomerular endothelial cells. Glomerulosclerosis was significantly reduced in MMP9-/- AN mice compared to that of WT, as demonstrated by GT and SR staining.
Conclusions: MMP-9 contributes to glomerulosclerosis at least in part by causing profibrotic changes in podocytes and glomerular endothelial cells.
Keywords: Matrix metalloproteinase 9; Glomerulosclerosis; Podocyte; Endothelial cell
Abbreviation
CKD: Chronic Kidney Disease; EMT: Epithelial-Mesenchymal Transition; EMT: Endothelial-Mesenchymal Transition; EndMT: Podocyte and Endothelial-Mesenchymal Transition; GBM: Glomerular Basement Membrane; VE-cadherin: Vascular Endothelial–Cadherin; MMP-9: Matrix Metalloproteinase 9; AN: Adriamycin Nephropathy; GFR: Glomerulus Filtration Rate; SR: Sirius Red; GT: Gomori Trichrome
Introduction
Glomerulosclerosis is a hallmark of Chronic Kidney Disease (CKD) [1,2]. It is characterized by the accumulation of myofibroblasts and excessive deposition of extracellular matrix components. Myofibroblasts are the key effectors in glomerulosclerosis. Both Epithelial-Mesenchymal Transition (EMT) of podocyte and endothelial-Mesenchymal Transition (EndMT) are major sources of myofibroblasts formation in kidney fibrosis [3,4].
Podocytes are terminally differentiated visceral epithelial cells that are critical components of the glomerular barrier and play an important role in selective permeability of the glomerular filtration barrier. Previous study indicated that podocyte damage leads to glomerulosclerosis [5]. Accumulating evidence indicates that in response to injurious stimuli, podocytes may undergo an EMT process, lose their epithelial surface markers such as P-cadherin, and express mesenchymal markers such as desmin. Podocytes were rendered motile after EMT, resulting in detachment from the Glomerular Basement Membrane (GBM) and podocyte loss, not apoptosis, and finally leading to a defective glomerular filtration, proteinuria and glomerulosclerosis [6].
Renal endothelial cells, especially glomerular endothelial cells, contribute to fibroblast formation in kidney by EndMT [7]. EndMT may be a notable source of activated fibroblasts or myofibroblasts [8]. During EndMT, endothelial cells eliminate endothelial markers, such as vascular endothelial–cadherin (VE-cadherin), and acquire mesenchymal markers, such as a-Smooth Muscle Actin (a-SMA) [9]. Previous studies have indicated that glomerular sclerosis were related to EndMT, and inhibition of EndMT could prevent glomerular sclerosis [10].
Matrix Metalloproteinase 9 (MMP-9) has been proven to cause kidney interstitial fibrosis [11]. However, whether it plays a role in glomerulosclerosis is not clear yet. We have demonstrated that MMP-9 induced EndMT in mouse peritubular endothelial cells downstream of TGF-β1 [11], indicating that it may also contribute to glomerulosclerosis. In the present study, we aim to investigate the role of MMP-9 in the development of glomerulosclerosis. This study hypothesized that MMP-9 may induce EMT process of podocytes and EndMT process of glomerular endothelial cells, thereby leading to glomerular sclerosis.
Materials and Methods
Animals and adriamycin-induced nephropathy model
Mouse Adriamycin Nephropathy (AN) was induced by a single injection of adriamycin (10.2 mg/kg, with physiological saline for controls) through tail vein in MMP-9 knockout (MMP-9-/-, BALB/c background) and wild-type control mice. All animals were sacrificed at 4 weeks after injection. Experiments were carried out in accordance with the protocols approved by the Animal Ethics Committee of Western Sydney Local Health District.
Urinary proteinuria concentration and kidney function
Urinary albumin to creatinine ratio was used to evaluate proteinuria. Urinary albumin concentration was determined by nephelometric method as reported [12]. Urinary creatinine concentration was determined by enzymatic method. Blood samples were taken from mice before sacrifice.
For calculated GFR detecting, mice were acclimatised to the metabolism cages for 48 h prior to 24 h urine collection. Urine samples were collected in metabolism cages 24 h before sacrifice. Serum creatinine levels were determined using the creatinine assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s instructions. Calculated GFR was evaluated by creatinine clearance using the standard formula.
Histological analysis
Four weeks after adriamycin treatment, paraffin-embedded kidney sections (4 μm) were deparaffinised with xylene and rehydrated through a descending ethanol gradient. Histological sections were examined following Sirius Red (SR) or Gomori Trichrome (GT) staining. Quantification of pulmonary and kidney fibrosis was performed as we described previously [13]. All scoring was performed in a blinded manner.
Immunofluorescence analysis
Frozen kidney blocks were cut into 7 μm sections and fixed with ice-cold acetone for 10 min at -20°C and blocked with 2% BSA for 1 h. Double immunofluorescence staining was performed using combinations of antibodies of P-cadherin and desmin, VE-cadherin and a-SMA, respectively. Tissue sections were then incubated with its corresponding fluorescence-conjugated secondary antibody. After washing with PBS, sections were counterstained with DAPI for 5 min before mounting with the fluorescence mounting medium. Images were obtained using a confocal microscope (Olympus FV1000) at ×40 magnification. For quantitative analysis, the percentage of the area stained positive for P-cadherin, desmin, or VE-cadherin, a-SMA, were counted on High Power Fields (HPFs) in a blinded manner.
Statistical analysis
Results were expressed as Mean ± SEM. Statistical significance was evaluated using unpaired two-tailed t-test for comparison between two groups. The level of significance was set at p<0.05.
Results
MMP-9 knockout reduces albuminuria and improves kidney function in mice
The urinary albumin/creatinine ratio and GFR were not different between wild-type control mice and MMP-9-/- controls. Adriamycin injection resulted in massive proteinuria in MMP-9 wild-type mice. However, it induced less pronounced albuminuria in MMP-9-/- mice (Figure 1A). Mice treated with adriamycin had markedly decreased GFR compared with normal animals. MMP-9-/- mice were protected from developing renal impairment, their GFR was significantly higher than wild-type AN mice (Figure 1B).
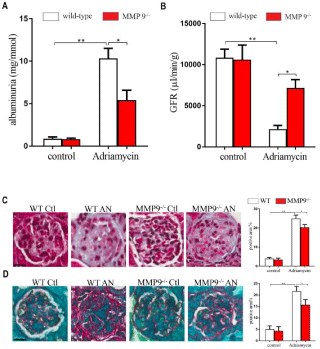
Figure 1: Effects of MMP-9 on albuminuria in mouse adriamycin
nephropathy. Mouse adriamycin nephropathy was induced by a single tail
vein injection of adriamycin (10.2 mg/kg) or saline as control in MMP-9-/- and wild-
type mice. (A) Quantitation of albuminuria, using unpaired two-tailed t
test and results are shown as ± SEM (n≥3 for each group). (B) Quantitation
of GFR, using unpaired two-tailed t test and results are shown as ± SEM
(n≥3 for each group). (C) Representative Gomori trichrome and (D) Sirius red
staining images were shown. Original magnification, X60. Scale bar, 40μm.
Quantitation of the glomerular area were shown, using unpaired two-tailed
t test. Results are shown as Mean ± SEM (n≥3 for each group). *P<0.05,
**P<0.01.
Effect of MMP-9 knockout on the progression of glomerulosclerosis
Kidney injury is characterized by glomerulosclerosis in AN. We showed here that in both wild-type and MMP-9-/- mice, adriamycin induced glomerular changes in contrast to the normal glomerular noticed in control mice. MMP-9-/- AN mice showed significantly less glomerulosclerosis than wild-type AN mice. These results indicate that knockout of MMP-9 gene attenuated the progression of glomerulosclerosis (Figure 1C and 1D).
Effect of MMP-9 knockout on podocytes and glomerular endothelial cells
Mesenchymal transition of podocytes (EMT) and endothelial cells (EndoMT) have been implicated in glomerulosclerosis [14,15]. We hypothesized that MMP-9 played a role in these is unknown. To test our hypothesis, the double staining of P-cadherin (i.e., podocyte marker) and desmin (i.e., podocyte injury marker), or VE-cadherin (i.e., endothelial marker) and a-SMA (i.e., myofibroblast marker) was performed for the kidney section. P-cadherin and desmin, VE-cadherin and a-SMA were not significantly different in wildtype controls compared with MMP-9-/- control kidneys (Figure 2A and 2C, 3A and 3C). Immunofluorescence staining exhibited that P-cadherin was primarily localized in the cell-cell junctional sites of the differentiated podocytes in wild-type and MMP-9-/- mice (Figure 2A and 2C). P-cadherin, a podocyte slit diaphragm marker, was significantly reduced, but as for desmin, a podocyte injury marker, was observably increased after adriamycin treatment in both wildtype and MMP-9-/- mice (Figure 2B and 2D). In wild-type mice and MMP-9-/- controls mice, VE-cadherin is tightly expressed in the whole glomerular tuft, but a-SMA existed along the capillary area (Figure 3A and 3C). In contrast, a-SMA was strongly localized in the whole glomerular tuft of AN mice (Figure 3B and 3D). Of note, MMP-9-/- AN significantly decreased the levels of desmin while increased P-cadherin (Figure 2D), and decreased a-SMA while increased VE-cadherin (Figure 3D) when compared with wild-type AN mice (Figure 2B and 3B). These results demonstrate that MMP- 9 contributes to EMT of podocytes and EndoMT that contribute to glomelularsclerogenesis.
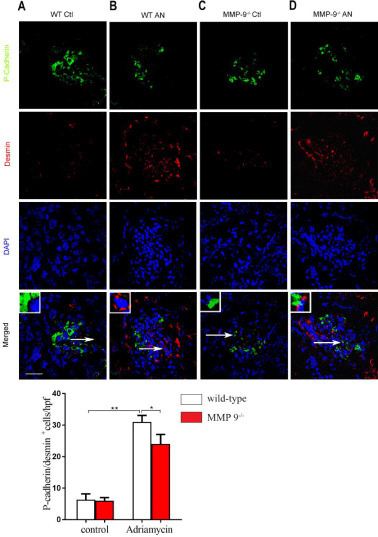
Figure 2: Effects of MMP-9 on podocyte P-cadherin and desmin distribution in
mouse adriamycin nephropathy. Mouse adriamycin nephropathy was induced
by a single tail vein injection of adriamycin (10.2 mg/kg) or saline as control
in MMP-9-/- and wild-
type mice. Representative immunofluorescence images
of P-cadherin (green color) and desmin (red color) and their glomerular colocalization
with DAPI (blue) were shown. Original magnification, X40.
Scale bar, 40μm. Quantitation of the area that are double-positive cells for
P-cadherin and desmin were counted on HPFs and used unpaired two-tailed
t test. Results are shown as Mean ± SEM (n=3 for each group). *P<0.05,
**P<0.01.
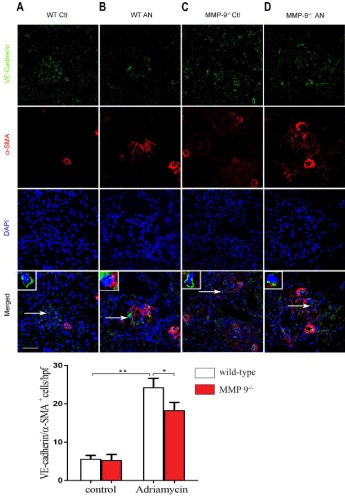
Figure 3: Effects of MMP-9 on endothelial cell VE-cadherin and a-SMA
distribution in mouse adriamycin nephropathy. Mouse adriamycin
nephropathy was induced by a single tail vein injection of adriamycin (10.2
mg/kg) or saline as control in MMP-9-/- and wild-
type mice. Representative
immunofluorescence images of VE-cadherin (green color) and a-SMA (red
color) and their glomerular co-localization with DAPI (blue) were shown.
Original magnification, X40. Scale bar, 40μm. Quantitation of the area that
are double-positive cells for VE-cadherin and a-SMA were counted on HPFs
and used unpaired two-tailed t test. Results are shown as Mean ± SEM (n≥3
for each group). *P<0.05, **P<0.01.
Discussion
Glomerulosclerosis is the final pathological process common to CKD [1]. MMP-9 plays a key role in kidney interstitial fibrosis [16], but little is known about its behaviour in glomerulosclerosis. In the present study, we investigated the effect of MMP-9 on glomerulosclerosis in murine AN, a model of human focal segmental glomerulosclerosis [17]. We found that glomerular fibrosis in MMP- 9-/- mice was less severe than wild-type controls combining with albuminuria and calculated GFR, indicating MMP-9 was involved in glomerulosclerosis.
Podocyte is regarded as a key player in glomerular health and disease [18]. Recent studies indicate that podocyte injury is a common trigger leading to the disruption of the filtration barrier and protein leakage [19], and ultimately result in glomerulosclerosis. Podocytes implicate in the glomerulosclerosis, at least in part, by EMT [20]. We examined the expression of P-cadherin, a predominant podocyte marker, and desmin, a mesenchymal marker, in glomeruli in AN mice. Our results showed that podocytes decrease expressing the P-cadherin protein and instead showed increased expression of desmin, which indicates that podocytes undergo EMT in both MMP- 9-/- and wild-type control AN mice. However, podocyte EMT was significantly decreased in MMP-9-/- mice, suggesting that MMP-9 plays an important role in podocyte EMT.
Emerging evidence indicate the critical role of EndMT in tissue fibrogenesis [21]. It is reported that EndMT of glomerular endothelial cells also involved in the glomerulosclerosis [22]. During EndMT, endothelial cells eliminate endothelial markers, such as Cluster of Differentiation 31 (CD31) and VE-cadherin, and acquire mesenchymal markers, such as fibroblast-specific protein 1 and a-SMA [9,10]. We showed here that AN injection increased VEcadherin expression, and decreased the expression of a-SMA in both MMP-9-/- mice and wild-type controls, suggesting that an EndoMT program is activated in glomerular endothelial cells after AN treatment. The EndoMT was more predominant in wild-type mice than MMP-9-/- ones, indicating that MMP-9 plays an important role in EndoMT of glomerular endothelial cells.
In conclusion, we found that MMP-9 plays an important role in kidney fibrosis, at least in part through podocyte EMT and glomerular endothelial cells EndMT. Pharmacological inhibition of the MMP- 9 could be a desirable therapeutic approach for treating glomerular renal disease.
References
- Djudjaj S, Boor P: Cellular and molecular mechanisms of kidney fibrosis. Molecular aspects of medicine 2019, 65: 16-36.
- Duffield JS: Cellular and molecular mechanisms in kidney fibrosis. The Journal of clinical investigation. 2014; 124: 2299-2306.
- Loeffler I, Wolf G. Epithelial-to-Mesenchymal Transition in Diabetic Nephropathy: Fact or Fiction? Cells. 2015; 4: 631-652.
- Sun YB, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation; research in biological diversity. 2016; 92: 102-107.
- Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. Journal of the American Society of Nephrology: JASN. 2005; 16: 2941-2952.
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010; 21: 212-222.
- Akis N, Madaio MP. Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney international. 2004; 65: 2223-2227.
- Kanasaki K, Shi S, Kanasaki M, He J, Nagai T, Nakamura Y, et al. Linagliptinmediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocininduced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014; 63: 2120-2131.
- Curci C, Castellano G, Stasi A, Divella C, Loverre A, Gigante M, et al. Endothelial-to-mesenchymal transition and renal fibrosis in ischaemia/ reperfusion injury are mediated by complement anaphylatoxins and Akt pathway. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association-European Renal Association. 2014; 29: 799-808.
- He J, Xu Y, Koya D, Kanasaki K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clinical and experimental nephrology. 2013; 17: 488-497.
- Zhao Y, Qiao X, Tan TK, Zhao H, Zhang Y, Liu L, et al. Matrix metalloproteinase 9-dependent Notch signaling contributes to kidney fibrosis through peritubular endothelial-mesenchymal transition. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2017; 32: 781-791.
- Miyazaki Y, Shimizu A, Pastan I, Taguchi K, Naganuma E, Suzuki T, et al. Keap1 inhibition attenuates glomerulosclerosis. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2014; 29: 783-791.
- Qiao X, Rao P, Zhang Y, Liu L, Pang M, Wang H, et al. Redirecting TGF-beta Signaling through the beta-Catenin/Foxo Complex Prevents Kidney Fibrosis. J Am Soc Nephrol. 2018; 29: 557-570.
- Wang X, Gao Y, Tian N, Wang T, Shi Y, Xu J, et al. Astragaloside IV inhibits glucose-induced epithelial-mesenchymal transition of podocytes through autophagy enhancement via the SIRT-NF-kappaB p65 axis. Scientific reports. 2019; 9: 323.
- Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J, et al. Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelialmesenchymal transition and fibrosis in CKD. Journal of the American Society of Nephrology: JASN. 2015; 26: 817-829.
- Zhao Y, Qiao X, Wang L, Tan TK, Zhao H, Zhang Y, et al. Matrix metalloproteinase 9 induces endothelial-mesenchymal transition via Notch activation in human kidney glomerular endothelial cells. BMC cell biology. 2016; 17: 21.
- Cao Q, Lu J, Li Q, Wang C, Wang XM, Lee VW, et al. CD103+ Dendritic Cells Elicit CD8+ T Cell Responses to Accelerate Kidney Injury in Adriamycin Nephropathy. Journal of the American Society of Nephrology: JASN. 2016; 27: 1344-1360.
- Maezawa Y, Onay T, Scott RP, Keir LS, Dimke H, Li C, et al. Loss of the podocyte-expressed transcription factor Tcf21/Pod1 results in podocyte differentiation defects and FSGS. Journal of the American Society of Nephrology: JASN. 2014; 25: 2459-2470.
- Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nature communications. 2017; 8: 413.
- Wu X, Gao Y, Xu L, Dang W, Yan H, Zou D, et al. Exosomes from high glucosetreated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Scientific reports. 2017; 7: 9371.
- Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. Journal of clinical medicine. 2016; 5.
- Lin JR, Zheng YJ, Zhang ZB, Shen WL, Li XD, Wei T, et al. Suppression of Endothelial-to-Mesenchymal Transition by SIRT (Sirtuin) 3 Alleviated the Development of Hypertensive Renal Injury. Hypertension. 2018; 72: 350-360.