
Review Article
Austin J Neurol Disord Epilepsy. 2014;1(2): 9.
The Role of Neuroimaging in Congenital Abnormalities of the Posterior Fossa
Poretti A1*, Wagner MW1 and Bosemani T1
1Section of Pediatric Neuroradiology, Division of Pediatric Radiology, The Russell H. Morgan Department of Radiology and Radiological Science, The Johns Hopkins University, USA
*Corresponding author: Poretti A, Section of Pediatric Neuroradiology, Division of Pediatric Radiology, The Russell H. Morgan Department of Radiology and Radiological Science, The Johns Hopkins University School of Medicine, Charlotte R. Bloomberg Children's Center, Sheikh Zayed Tower, Room 4174, 1800 Orleans Street, Baltimore, MD 21287-0842, USA
Received: August 18, 2014; Accepted: November 29, 2014; Published: December 01, 2014
Abstract
The evaluation of the posterior fossa has gained significant pace and importance in the last 20 years based on the successful progress in neuroimaging. Nowadays, conventional and advanced neuroimaging techniques allow a detailed evaluation of the complex anatomical structures within the compact posterior fossa. A wide spectrum of congenital abnormalities including malformations and disruptions has been shown. Neuroimaging is mandatory for the diagnosis of congenital abnormalities of the posterior fossa. Welldefined diagnostic criteria based on neuroimaging findings are available for the different disorders. Familiarity with the spectrum of congenital posterior fossa anomalies and their diagnostic criteria is mandatory for an accurate evaluation. An accurate and up-to-date classification is important for therapy, prognosis, and genetic counseling of the affected children and their families. In addition, neuroimaging provides useful information in elucidating the pathogenesis, establishing a phenotype (neuroimaging-) genotype correlation, and predicting the neurological outcome.
Keywords: Neuroimaging; Children; Cerebellum; Malformation; Disruption
Introduction
In the last few decades, progress in neuroimaging techniques, genetic analysis and mouse model research has led to a significant improvement in the definition of congenital cerebellar abnormalities and henceforth a better understanding of their pathogenesis. New classifications of congenital posterior fossa abnormalities have been proposed based on molecular genetics and developmental biology [1,2].
Neuroimaging plays a key role in the diagnosis of congenital brain abnormalities. An accurate diagnosis of these complex abnormalities is important for three primary reasons: 1) to determine inheritance pattern and risk of recurrence, 2) to evaluate for multisystem involvement (e.g. kidneys and liver), and 3) to provide prognostic implications for the child and family. The diagnosis should include the differentiation between inherited (genetic) and acquired (disruptive) abnormalities. A Malformation is defined as a congenital morphologic anomaly of a single organ or body part due to an alteration of the primary developmental program caused by a genetic defect [3]. Gene mutations causing malformations may be "de novo" or be inherited following different patterns that imply a different recurrence risk for further offspring. A Disruption is defined as a congenital morphologic anomaly due to the breakdown of a body structure that had a normal developmental potential [3]. Disruptions may be caused by e.g. prenatal infection, hemorrhage, or ischemia and commonly involve the cerebellum [4]. Disruptions are acquired lesions with very low recurrence risk. However, a genetic predisposition to disruptive lesions may be present. Dominant mutations in COL4A1 lead to change in the basal membrane of capillaries resulting in microangiopathy [5]. Within the brain, the microangiopathy may lead to hemorrhage and/or ischemia and result in porencephaly or unilateral cerebellar hypoplasia [6,7]. Homozygous mutations in NED1 have been shown to cause severe microcephaly, agenesis of the corpus callosum, scalp rugae, and the fetal brain disruption-like phenotype [8].
In addition, neuroimaging findings may: 1) allow the definition of sub-phenotypes within a group of posterior fossa malformations, 2) establish correlations between the neuroimaging phenotype and genotype, and 3) facilitate a more targeted genetic analysis. Moreover, the application of conventional and, particularly, advanced Magnetic Resonance Imaging (MRI) sequences may better define the etiology and pathogenesis of congenital posterior fossa abnormalities. Diffusion Tensor Imaging (DTI) provides detailed qualitative and quantitative information about micro-structure and organization of the white matter tracts [9]. On DTI and fiber tractography images, white matter tracts with an abnormal course, failure to decussate, or in an ectopic location are suggestive of axonal guidance disorders [9]. Recently, DTI allowed the delineation of possible two new axonal guidance disorders in single patients [10,11]. Susceptibility- Weighted Imaging (SWI) is highly sensitive for blood, blood products and calcifications and may be helpful in supporting a disruptive pathomechanism [12]. Finally, neuroimaging findings may predict the neurological outcome. The involvement of selected anatomical regions on neuroimaging may serve as biomarkers of cognitive functions and behavior.
In this review article, we will discuss the role of neuroimaging in selected congenital abnormalities of the posterior fossa including Dandy-Walker Malformation (DWM), Joubert Syndrome (JS), Rhombencephalosynapsis (RES), Pontine Tegmental Cap Dysplasia (PTCD), and Unilateral Cerebellar Hypoplasia (UCH) (Table 1).
![]()
Disease
Diagnostic finding
Cerebellar vermis
IV. ventricle
Posterior fossa
Hydrocephalus
DWM
Cystic dilation of the IV. ventricle + vermian hypoplasia + elevation
Hypoplastic
Enlarged
Enlarged
Majority of the patients
JS
Molar tooth sign
Hypoplastic / dysplastic
Mostly enlarged + upwards displaced
Variable
Rare
RES
Continuity of the cerebellar hemispheres
Completely or
partially absent
Key-hole shape
Normal
50% of the patients
PTCD
Tegmental cap
Hypoplastic /
dysplastic
Reduced
Normal
Rare
UCH
Absence of one cerebellar hemisphere
Normal or
hypoplastic
Normal
Variable
Rare
Table 1: Distinguishing neuroimaging findings in selected congenital abnormalities of the posterior fossa.
Dandy-Walker malformation
Dandy-Walker Malformation (DWM) is the most prevalent human cerebellar malformation (about 1:30.000 live births) [13]. The vast majority of patients with DWM are sporadic without familial recurrence or association with consanguinity. The overall empiric recurrence risk is accordingly low (1-5%). The etiology of DWM is still poorly understood and most likely multifactorial. DWM is due to a genetic cause in the majority of patients however. DWM may occur isolated or be part of a well-defined Mendelian disorder such as 1) Ritscher-Schinzel syndrome or cranio-cerebello-cardiac syndrome (OMIM 220210) secondary to mutations in KIAA0196 [14,15] or 2) PHACE(S) syndrome (OMIM 606519) [16,17]. In addition, DWM has been reported in association with various chromosomal anomalies which include trisomy 9, 13, 18, triploidy, tetrasomy, and several other partial duplications and deletions [18]. Finally, mutations in six genes (ZIC1, ZIC4, FOXC1, FGF17, LAMC1, and NID1) have been found in few patients with DWM [19-22]. The function of these genes suggests that DWM may result from a complex disruption of the interaction between the developing cerebellum and the developing posterior fossa mesenchyme with its derivates.
The majority of children with DWM present in the first year of life secondary to symptoms and signs of increased intracranial pressure [23]. Macrocephaly is the most common clinical manifestation affecting 90-100% of children during the first months of life [24]. Nowadays, DWM is increasingly diagnosed prenatally. The outcome of children with DWM is variable. Overall, at least one third of DWM patients have normal cognitive functions [25-27]. In DWM associated with Mendelian syndromes or chromosomal abnormalities, the presence of systemic involvement (e.g. cardiovascular, urogenital, and skeletal anomalies) may also influence the prognosis.
In DWM, neuroimaging findings: 1) are required for the diagnosis, 2) allow differentiation from other cystic malformations of the posterior fossa, and 3) may predict cognitive outcome. The key neuroimaging features of DWM are 1) hypoplasia (or rarely agenesis), elevation, and counterclockwise rotation of the cerebellar vermis and 2) cystic dilatation of the fourth ventricle which extends posteriorly filling out nearly the entire posterior fossa (Figure 1) [28]. Both findings are consistently present and required for the diagnosis. The cerebellar hemispheres are typically displaced rostrally and laterally, and their volume is often reduced. The brainstem may be hypoplastic. The posterior fossa is usually enlarged., The tentorium, torcular and transverse sinuses are commonly elevated, and hydrocephalus may be present. In 30-50% of patients, DWM is associated with additional malformations including dysgenesis/agenesis of the corpus callosum, occipital encephalocele, polymicrogyria, and heterotopia [28].
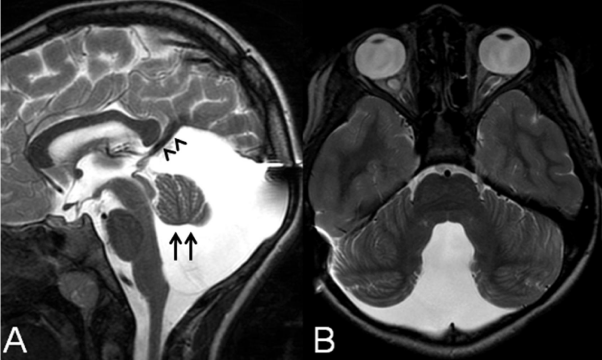
Figure 1: A: Midsagittal T2-weighted image in a 10-year-old girl with Dandy-Walker malformation after ventriculo-peritoneal and cysto-peritoneal shunt in the first year of life shows hypoplasia of the cerebellar vermis which is elevated and rotated upwards (arrows), cystic dilatation of the fourth ventricle extending posteriorly filling out nearly the entire enlarged posterior fossa, and elevation of the tentorium and torcula. B: Axial T2-weighted image of the same child shows antero-lateral displacement of the cerebellar hemispheres which are mildly hypoplastic (both images reprinted with permission from Poretti A, Millen KJ and Boltshauser E, Dandy-Walker malformation, in: Boltshauser E and Schmahmann, JD., ed. Cerebellar disorders in children. London: Mac Keith Press, 2012; pp. 140-148).
Neuroimaging allows differentiating DWM form other cystic posterior fossa malformations such as Blake's pouch cyst, posterior fossa arachnoid cysts, and mega cisterna magna [29,30]. A Blake's pouch cyst is caused by lack of fenestration of the Blake's pouch resulting in absence of communication between the fourth ventricle and subarachnoid space [29-31]. The typical neuroimaging findings include the presence of a cyst that lies in a retrocerebellar or infra-retrocerebellar location and directly communicates with the fourth ventricle, which is usually enlarged. The choroid plexus is displaced along the antero-superior aspect of the cyst and inferior to the vermis, and is best visualized in a sagittal T1-weighted post-contrast sequence as an enhancing structure [30]. The absence of communication between the fourth ventricle and subarachnoid space results in a tetraventricular hydrocephalus. Mild mass effect from the infra-retrocerebellar cyst may result in indentation of the inferior vermis or the caudal and medial aspect of the cerebellar hemispheres, but the size and shape of the cerebellum is globally normal. The posterior fossa is typically normal in size. Supratentorial morphological abnormalities other than hydrocephalus are usually absent. Arachnoid cysts are duplications of the arachnoid membrane that produce fluid-filled cysts. 10% of pediatric arachnoid cysts occur in the posterior fossa and may be located inferior or posterior to the vermis in a midsagittal location (retrocerebellar), cranial to the vermis in the tentorial hiatus (supravermian), anterior or lateral to the cerebellar hemispheres, or anterior to the brainstem [32]. Neuroimaging demonstrates a well-circumscribed extra-axial fluid collection or cyst which is isointense to Cerebrospinal Fluid (CSF) on all pulse sequences. The presence of proteinaceous content may lead to lack of complete signal suppression on a Fluid Attenuation Inversion Recovery (FLAIR) sequence. DTI reveals free water motion or facilitated diffusion similar to CSF. The cyst walls are usually too thin to be visualized by MRI. Arachnoid cysts do not communicate with the fourth ventricle or subarachnoid space. Arachnoid cysts may enlarge during infancy and produce mass effect on the cerebellum and vermis, which may cause a secondary obstruction of the ventricular system, hydrocephalus and/or remodeling/thinning of the overlying occipital bone. Mega cisterna magna is an enlarged cisterna magna (=10mm diameter on midsagittal images) with an intact vermis, normal fourth ventricle, and in some patients an enlarged posterior fossa [29,33,34]. Mega cisterna magna freely communicates with the fourth ventricle and cervical subarachnoid space, which is confirmed on CSF flow studies and results in consistent absence of hydrocephalus [34]. Finally, isolated inferior vermis hypoplasia should be differentiated from DWM. Isolated inferior vermis hypoplasia is defined by partial absence of the inferior portion of the cerebellar vermis, which is best demonstrated in the midsagittal images. The rest of the vermis, cerebellar hemispheres, fourth ventricle, and posterior fossa have a normal size and architecture. In the literature, there is some confusion about isolated inferior vermian hypoplasia and some authors call it "Dandy-Walker variant" [35]. In addition, some patients show only a partial neuroimaging phenotype of DWM. To classify malformations that do not fulfill the criteria of a "true" DWM, other terms such as "Dandy-Walker complex" or "Dandy-Walker spectrum" have been introduced. These terms ("Dandy-Walker variant", "Dandy- Walker complex", and "Dandy-Walker spectrum") have been used by different authors with variable findings. This has led to a lack of specificity with regards to the definition of these terms and confusion over "true"' DWM. If neuroimaging diagnostic criteria do not allow to distinguish DWM from other posterior fossa malformations, a detailed anatomical description (e.g. inferior cerebellar vermis or global cerebellar hypoplasia) is highly recommended and preferable over non-specific terms such as "Dandy-Walker variant", which should be avoided or even better abandoned [28].
Neuroimaging findings of DWM have been shown to correlate with cognitive outcome. Normal lobulation of the cerebellar vermis and absence of associated brain abnormalities seem to be neuroimaging biomarkers of normal cognitive functions in DWM [26]. Boddaert et al assessed cognitive functions in 20 children with DWM and correlated them with neuroimaging findings [36]. The authors divided the patients in two groups based on cognitive outcome (normal vs. impaired). While all patients with normal cognitive functions had normal lobulation of the cerebellar vermis and no supratentorial anomalies, three of the six children with intellectual disability had abnormal vermis lobulation and dysgenesis of the corpus callosum. Cognitive functions were normal in 82% of children with normal vermis lobulation and abnormal in all children with abnormal vermis lobulation. Similar results have been reported in a study including 26 children with DWM [37]. These studies suggest that impaired cognitive functions in DWM result most likely from the cerebellar abnormality and not from increased intracranial pressure. In addition, the presence of supratentorial abnormalities may negatively affect cognitive functions [23, 26, 36-38].
Joubert syndrome
Joubert Syndrome (JS) is a rare midbrain-hindbrain malformation with an estimated prevalence between 1:80,000 and 1:100,000 live births [39]. In almost all patients, JS is inherited with an autosomal recessive pattern and has a 25% recurrence risk in the affected families. Only mutations in OFD1 are exceptional and inherited with an X-linked pattern. Twenty-seven genes have been associated with JS so far [40-45]. All genes encode for proteins that localize to primary non-motile cilia and its basal body which play a key role in the development and functioning of the brain, retina, kidney, liver and other organs [46]. Primary cilia mediate various signaling processes and brain malformations in JS and may result from defects in midline fusion of the developing vermis or defects in sonic hedgehog (Shh)- mediated granule cell proliferation [47,48]. In addition, cilia have been shown to have a repressing role onto the Wnt signaling by maintaining a discrete range of Wnt responsiveness [47]. Both Wnt and Shh signaling have been linked to axonal guidance.
About 30-40% of children with JS present during the neonatal period because of irregular breathing pattern, which include phases of apnea and tachypnea. Later in life, children with JS present with muscular hypotonia, cerebellar ataxia and ocular motor apraxia [40]. Cognitive functions are impaired in almost all patients, but the degree of impairment ranges between profound and mild [49]. Recently, horizontal head titubation has been reported as a new, early (presentation within the first 2 months of age), and benign finding of JS in 13 young affected children [50]. In these children, head titubation was present only when children were awake and decreased in severity over time until spontaneous resolution. Systemic involvement may be present and includes renal (nephronophthisis 25%), eye (retinal dystrophy 30% and colobomas 20%), liver (congenital hepatic fibrosis 15%), and skeletal (different forms of polydactyly 20%) abnormalities [40]. Renal and liver involvement may cause high morbidity and mortality and needs appropriate work-up and regular follow-up. Based on the systemic involvement, 6 phenotypes have been described: 1) pure JS (purely neurological involvement), 2) JS with eye involvement, 3) JS with kidney involvement, 4) JS with involvement of eyes and kidneys, 5) JS with liver involvement and 6) JS with oral-facial-digital involvement or the oral-facial-digital syndrome type VI (OFDVI, OMIM: 277170) [40]. The presence of tongue hamartoma, additional frenula, upper lip notch, mesoaxial polydactyly of one or more hands or feet and/or hypothalamic hamartoma differentiates OFDVI from the other phenotypes [51]. Some degree of correlation between these clinical phenotypes and the genotype has been shown. The strongest correlation is between mutations in TMEM67 and liver involvement [52].
In JS, neuroimaging findings: 1) are required for the diagnosis, 2) show a weak correlation with the genotype, 3) provide information about the possible pathogenesis, and 4) do not correlate with the neurological outcome. The so-called Molar Tooth Sign (MTS) is the pathognomonic neuro-anatomical and neuroimaging feature of JS [53-55]. The MTS is characterized by thickened, elongated, and horizontally orientated Superior Cerebellar Peduncles (SCP) and an abnormally deep interpeduncular fossa [53-55]. The spectrum of neuroimaging findings in JS is, however, beyond the molar tooth sign and vermian hypoplasia and dysplasia, confirming the heterogeneity in Joubert syndrome not only from the clinical and genetic, but also from the neuroimaging point of view [55]. The degree of vermian hypoplasia, size and morphology of the cerebellar hemispheres, form of the MTS, size of the posterior fossa, and shape and size of the cerebellar hemispheres are variable. Morphological abnormalities of the brainstem are present in about 30% of the patients. Supratentorial involvement occurs in about 30% of the patients and includes callosal dysgenesis, cephaloceles, hippocampal malrotation, migrational disorders, and ventriculomegaly. Differences in neuroimaging findings can be present in siblings representing intrafamilial heterogeneity.
A weak correlation between neuroimaging findings and genotype has been suggested. In children with JS due to deletions in NPHP1, the MTS may appear to be less striking with less extensive cerebellar vermis hypoplasia, and an elongated but not thickened SCP [56]. Mutations in KIF7, one of the genes associated with JS, have also been found in children with acrocallosal syndrome [57-59]. Acrocallosal syndrome is characterized by agenesis or dysgenesis of the corpus callosum, craniofacial dysmorphism, duplication of the hallux, postaxial polydactyly, and severe intellectual disability [59]. Accordingly, it is possible that KIF7 may be the major gene in JS patients with agenesis or dysgenesis of the corpus callosum. Finally, hypothalamic hamartoma have been reported only in JS patients with the OFDVI phenotype [51]. Recently, C5orf42 has been shown to be the major gene associated with the OFDVI phenotype [60]. Accordingly, the presence of hypothalamic hamartoma may correlate with mutations in C5orf42.
In JS, DTI and fiber tractography shows absence of decussation of both the SCP and Corticospinal Tracts (CST) at the level of the ponto-mesencephalic junction and lower brainstem, respectively [61,62]. The DTI and tractography findings match previous neuropathological reports by showing an almost complete absence of decussation of both SCP and CST [63,64]. In addition, it has been shown that AHI1 mRNA (AHI1 is one the genes associated with JS) is expressed in the cell bodies that give rise to axonal tracts (CST and SCP) which fail to decussate in JS [65]. Absence of decussation of SCP and CST suggests an underlying defect in axonal guidance in JS [66].
Rhombencephalosynapsis
Rhombencephalosynapsis (RES) is a rare, sporadic cerebellar malformation that is thought to be caused by aberrant dorsal-ventral patterning [67,68]. The sporadic nature attributes to a low recurrence risk. The majority of children with RES do not have other syndromic findings. RES is however a key feature of Gómez-López-Hernández syndrome (OMIM 601853). The Gómez-López-Hernández syndrome is defined by the presence of RES and bilateral parietal alopecia [69,70]. In addition, trigeminal anesthesia causing corneal opacities and craniofacial dysmorphisms such as midface hypoplasia, low-set posteriorly rotated ears, brachycephaly, and hypertelorism may be present. Finally, features of VACTERL association (Vertebral anomalies, Anal atresia, Cardiovascular anomalies, Trachea- Esophageal fistula, Renal anomalies, Limb defects) are seen in some patients with RES [71].
Children with RES may present with truncal and limb ataxia, hypotonia, abnormal eye movements such as strabismus and nystagmus, and delayed motor development in the first years of life [67,72]. About 85% of patients have head-shaking stereotypies (repetitive Figure 2-5 or side-to-side swinging motion) that may represent a response to deficits in central vestibular processing [73]. In some patients, the diagnosis of RES is made during the neonatal period because of the association with hydrocephalus [68,74]. Long-term cognitive outcome varies between normal functions and severe impairment [72]. Some degree of intellectual disability is present in the majority of patients. In addition, behavioral problems such as attention deficits may be present. Systemic involvement is uncommon.
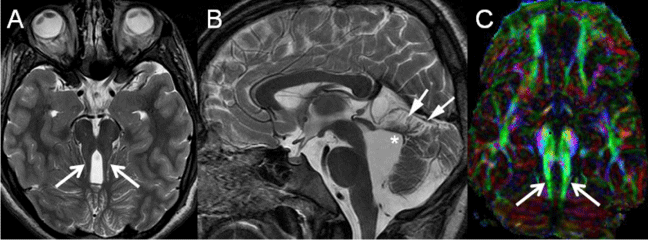
Figure 2: A: Axial T2-weighted image of a child with Joubert syndrome shows the molar tooth sign with elongated, thickened and horizontally orientated superior cerebellar peduncles (arrows) and a deepened interpeduncular fossa. B: Midsagittal T2-weighted image of the same patient demonstrates severe hypoplasia of the cerebellar vermis with dysplasia of the vermian remnants (arrows), shortening of the ponto-mesencephalic isthmus and enlargement of the fourth ventricle with upwards displacement of the fastigium (asterisk). C: Axial color-coded fractional anisotropy maps (DTI) of the same patient reveals the superior cerebellar peduncles (arrows) as encoded in green (green = anterior ⇔ posterior orientation), while physiologically the superior cerebellar peduncles are encoded in blue (blue = superior ⇔ inferior orientation); additionally, the midline "red dot" (red = right ⇔ left orientation) is missing representing absence of decussation of the superior cerebellar peduncles (images A and B reprinted with permission from Poretti A et al, Am J Med Genet C Semin Med Genet, 2014;166C:211-226; image C reprinted with permission from Poretti A et al, AJNR Am J Neuroradiol, 2007;28:1929-33).
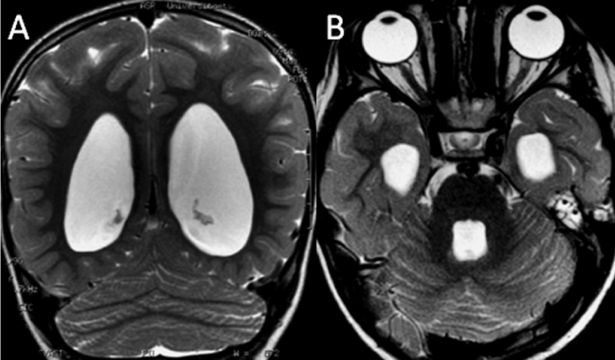
Figure 3: A: Posterior coronal T2-weighted image of a girl with rhombencephalosynapsis demonstrates continuity of the cerebellar hemispheres with an abnormal, transverse orientation of cerebellar folia and ventriculomegaly. B: Axial T2-weighted image of the same patient reveals continuity of the cerebellar hemispheres without an intervening vermis and dilatation of the temporal horns of the lateral ventricles (both images reprinted with permission from Poretti A et al, Eur J Paediatr Neurol, 2009;13:28-33).
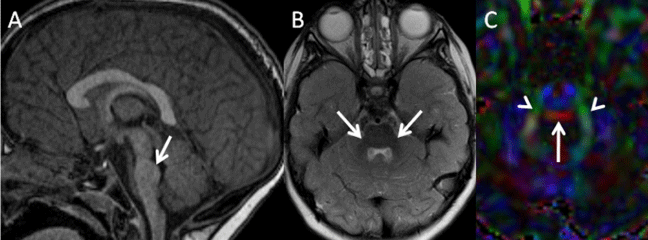
Figure 4: A: Midsagittal T1-weighted image of a 3-year-old girl with pontine tegmetal cap dysplasia reveals a flat ventral pons and a cap covering dorsal pons (arrow) and protruding into the fourth ventricle. B: Axial T2-weighted image of the same child demonstrates mild cerebellar hypoplasia with hypoplastic middle cerebellar peduncles (arrows). C: Axial color-coded fractional anisotropy map (DTI) of the same patient shows an ectopic band of fibers in red (horizontal orientation) at the dorsal aspect of the pons (arrow) and small middle cerebellar peduncles in green (arrowheads anterior ⇔ posterior orientation) (all images reprinted with permission from Poretti A et al, Am J Med Genet C Semin Med Genet, 2014;166C: 211-226).
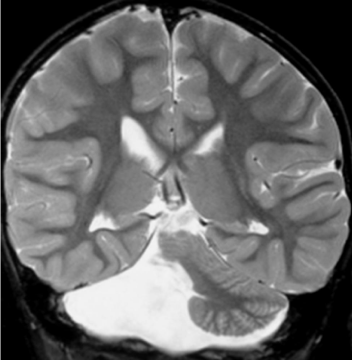
Figure 5: Unilateral cerebellar hypoplasia. A. Coronal T2-weighted image shows a total aplasia of the right cerebellar hemisphere in a 4-year-old child found in investigation of developmental delay (reprinted with permission from Poretti A et al, Eur J Paediatr Neurol, 2009; 13: 397-407).
In RES, neuroimaging findings: 1) are required for the diagnosis and 2) correlate with the neurological outcome. The key neuroimaging findings in RES are complete or partial absence of the cerebellar vermis and continuity of the cerebellar hemispheres, superior cerebellar peduncles, and dentate nuclei, which arch in a horseshoe shape across the midline, resulting in a keyhole-shaped fourth ventricle [67,68,72]. The posterior coronal sections are crucial to evaluate the horizontal cerebellar folial pattern. On the midsagittal images, the dentate nuclei are seen rather than the vermis (if normal anatomy is present, the vermis separates the dentate nuclei in the midline). Only in the most severe cases, the entire cerebellar volume is reduced. RES is often associated with midbrain abnormalities such as midline fusion of the colliculi [68]. Hydrocephalus is present in about 50% of the patients and aqueductal stenosis is the most common cause of hydrocephalus in RES (about 60% of RES with hydrocephalus) [68,74]. Finally, supratentorial malformations may be associated with RES including dysgenesis of the corpus callosum, absence of the septum pellucidum, absent olfactory bulbs, abnormal dysplastic temporal cortex, and, in rare patients, holoprosencephaly [68]. In RES, DTI 1) confirms the absence of transversely oriented fibers in the midsection of the cerebellum, 2) demonstrates the vertical direction of the fibers in the midportion of the fused cerebellum, and 3) shows that the deep cerebellar nuclei appear more closely approximated [61].
Neuroimaging findings correlate with the neurological outcome of patients with RES. A poor neurodevelopmental outcome in children with RES has shown to be associated with the severity of RES (children with complete agenesis of the vermis had a poorer outcome compared to children with partial agenesis), severity of ventriculomegaly, and presence of aqueductal stenosis, fused colliculi, abnormal temporal cortex, and holoprosencephaly [67, 68].
Pontine tegmental cap dysplasia
Pontine Tegmental Cap Dysplasia (PTCD) is a rare, sporadic posterior fossa malformation with unknown genotype and no familial recurrence and is thought to belong to the group of axonal guidance disorders [66,75,76]. PTCD is characterized by multiple cranial nerve involvement. The most commonly affected cranial nerves are the vestibulocochlear, facial, trigeminal, and glossopharyngeal nerves with resultant bilateral sensory deafness, bilateral trigeminal anesthesia causing corneal ulcers, bilateral facial paralysis, and difficulty in swallowing needing gastrostomy in some children [75- 77]. In addition, ocular movement disorders such as nystagmus and cerebellar signs such as truncal and limb ataxia and dysarthria are common in PTCD [75-77]. Finally, global developmental delay and intellectual disability are features of almost all children with PTCD and range between mild cognitive delay and severe disability [75- 78]. Systemic involvement with vertebral segmentation anomalies, rib malformations, and congenital heart defects has been observed in several patients [75-77, 79].
In PTCD, neuroimaging findings: 1) are required for the diagnosis, 2) suggest the possible pathogenesis, and 3) correlate with the neurological outcome. PTCD has a distinct neuroimaging pattern including a flattened ventral pons, an abnormal curved structure (the so called "cap") covering the middle third of the pontine tegmentum and projecting into the fourth ventricle, hypoplasia and dysplasia of the cerebellar vermis, slightly elongated and horizontally orientated SCP remotely reminiscent of a "molar tooth like" appearance, thin to almost absent Middle Cerebellar Peduncles (MCP), absence of the Inferior Cerebellar Peduncles (ICP), a short ponto-mesencephalic isthmus, an altered shape of the lower brainstem due to hypoplasia or agenesis of the inferior olivary nuclei, and a slightly enlarged fourth ventricle [75-77]. Additional neuroimaging findings include hypoplastic or absent facial and cochlear nerves, duplicated internal auditory canals, and, in few patients, supratentorial abnormalities such as dysgenesis of the corpus callosum and mild ventriculomegaly [80].
In PTCD, DTI shows the absence of the transverse pontine fibers and presence of an abnormal bundle of ectopic, transverse fibers at the level of the "cap" in the dorsal pons [75,76]. In addition, DTI revealed absence of the decussation of the SCP at the ponto-mesencephalic junction [76]. Finally, Caan et al recently reported extra-axial arcuate tracts connecting the basal pons to the cerebellar hemispheres and bypassing the MCP [81]. Both semi-arcs fused in a single midline trunk, caudally connecting to the region of the internal arcuate fibers in the medulla oblongata. Absence of transverse pontine fibers, ectopic transverse fibers in the dorsal pons, absence of decussation of the SCP, and ectopic prepontine arcuate fibers are highly suggestive of abnormal axonal guidance and/or neuronal migration. In addition, DTI shows that long tracts correlating reasonably well with the location and pathway of the CST were smaller when compared with those of healthy subjects [76]. The reduction in size of these tracts may be the cause of mirror movements of hands and feet as shown in a 5-year-old girl with PTCD [79]. The presence of mirror movements further supports an axonal guidance disorder as the cause of PTCD [66].
Neuroimaging findings correlate with the neurological outcome of patients with PTCD. The degree of brainstem dysplasia seems to correlate with the developmental disability: mildly affected patients tend to have a rounded bump (the so-called cap) and those who are more severely affected tend to have a more angular brainstem kink (a so-called beak) [2].
Unilateral cerebellar hypoplasia
Unilateral Cerebellar Hypoplasia (UCH) is a relatively rare neuroimaging finding encompassing a spectrum ranging from complete aplasia (absence of one cerebellar hemisphere) to mild asymmetry in the size of the cerebellar hemispheres [82,83]. Minor asymmetry in the size of the cerebellar hemispheres is occasionally seen as an incidental finding without clinical significance, whereas severe UCH is expected to be of clinical relevance. Increasing experience with prenatal ultrasound and fetal MRI has proved that UCH is of prenatal (representing a disruption) origin, with hemorrhage contributing to the leading fetal cause [4,83]. This is important for genetic counseling because UCH has a low recurrence risk. However, a genetic predisposition to disruption such as mutations in COL4A1 may be present [6]. Prenatal cerebellar hemorrhages originate mostly in the subpial external granular layer, a germinal matrix which is thickest at 24 weeks of gestation and begins to involute at 30 weeks of gestation [84]. Accordingly, the majority of reported prenatal cerebellar hemorrhages have been detected between 18 and 24 weeks of gestation [85-91]. Follow-up examinations after fetal cerebellar hemorrhages have shown a gradual reduction in size of the affected cerebellar hemisphere consistent with UCH [85,90]. UCH may occur isolated, or be associated with other disruptive lesions such as schizencephaly, or as part of PHACE(S) syndrome [17,92]. UCH with or without involvement of the cerebellar vermis is present in about 75% of PHACE(S) patients with posterior fossa involvement, while its association with DWM is rare (<5% of patients). In PHACE(S), UCH is almost always associated with abnormalities of the ipsilateral internal carotid artery or a persistent embryonic carotid-basilar connection [17]. This association further suggests a disruptive origin.
The most common clinical findings are developmental and speech delay, hypotonia, ataxia, and abnormal ocular movements [82,83,93]. The cognitive outcome is variable, ranging from almost normal development to marked motor and intellectual disability.
In UCH, neuroimaging findings: 1) are required for the diagnosis and 2) correlate with the neurological outcome. In UCH, there is variable involvement and volume loss in the cerebellar hemisphere and vermis [82,83,93]. In the hypoplastic hemisphere, abnormal foliation and/or clefts may be present. The volume of the posterior fossa may be variable. In almost all children, brainstem asymmetry is also observed. Additional supratentorial disruptive lesions such as porencephalic cysts or clefts may be present and support the disruptive origin of unilateral cerebellar hypoplasia. Evidence of hemorrhage may be present and is best visualized on SWI sequences. The absence of hemosiderin, however, does not exclude hemorrhage since the blood brain barrier is permeable to hemosiderin laden macrophages between 24-32 weeks of gestation.
In UCH, neuroimaging findings may correlate with the neurological outcome. Involvement of the cerebellar vermis is often, but not consistently associated with a poor cognitive outcome, whereas an intact cerebellar vermis is associated with normal outcome and no truncal ataxia [83].
Conclusion
Neuroimaging evaluation is mandatory for the diagnosis of congenital posterior fossa abnormalities. In addition, neuroimaging provides useful information in elucidating the pathogenesis, establishing a phenotype (neuroimaging-) genotype correlation, and predicting the neurological outcome.
References
- Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012; 135: 1348-1369.
- Doherty D, Millen KJ, Barkovich AJ. Midbrain and hindbrain malformations: advances in clinical diagnosis, imaging, and genetics. Lancet Neurol. 2013; 12: 381-393.
- Hennekam RC, Biesecker LG, Allanson JE, Hall JG, Opitz JM, Temple IK, et al Elements of Morphology Consortium. Elements of morphology: general terms for congenital anomalies. Am J Med Genet A. 2013; 161: 2726-2733.
- Poretti A, Prayer D, Boltshauser E. Morphological spectrum of prenatal cerebellar disruptions. Eur J Paediatr Neurol. 2009; 13: 397-407.
- Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005; 308: 1167-1171.
- Vermeulen RJ, Peeters-Scholte C, Van Vugt JJ, Barkhof F, Rizzu P, van der Schoor SR, et al. Fetal origin of brain damage in 2 infants with a COL4A1 mutation: fetal and neonatal MRI. Neuropediatrics. 2011; 42: 1-3.
- Yoneda Y, Haginoya K, Kato M, Osaka H, Yokochi K, Arai H, et al. Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly. Ann Neurol. 2013; 73: 48-57.
- Paciorkowski AR, Keppler-Noreuil K, Robinson L, Sullivan C, Sajan S, Christian SL, et al. Deletion 16p13.11 uncovers NDE1 mutations on the non-deleted homolog and extends the spectrum of severe microcephaly to include fetal brain disruption. Am J Med Genet A. 2013; 161: 1523-1530.
- Poretti A, Meoded A, Rossi A, Raybaud C, Huisman TA. Diffusion tensor imaging and fiber tractography in brain malformations. Pediatr Radiol. 2013; 43: 28-54.
- Irahara K, Saito Y, Sugai K, Nakagawa E, Saito T, Komaki H, et al. Pontine malformation, undecussated pyramidal tracts, and regional polymicrogyria: a new syndrome. Pediatr Neurol. 2014; 50: 384-388.
- Kweldam CF, Gwynn H, Vashist A, Hoon AH, Huisman TA, Poretti A. Undecussated superior cerebellar peduncles and absence of the dorsal transverse pontine fibers: a new axonal guidance disorder? Cerebellum. 2014; 13: 536-540.
- Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014; 40: 530-544.
- Osenbach RK, Menezes AH. Diagnosis and management of the Dandy-Walker malformation: 30 years of experience. Pediatr Neurosurg. 1992; 18: 179-189.
- Leonardi ML, Pai GS, Wilkes B, Lebel RR. Ritscher-Schinzel cranio-cerebello-cardiac (3C) syndrome: report of four new cases and review. Am J Med Genet. 2001; 102: 237-242.
- Elliott AM, Simard LR, Coghlan G, Chudley AE, Chodirker BN, Greenberg CR, et al. A novel mutation in KIAA0196: identification of a gene involved in Ritscher-Schinzel/3C syndrome in a First Nations cohort. J Med Genet. 2013; 50: 819-822.
- Rossi A, Bava GL, Biancheri R, Tortori-Donati P. Posterior fossa and arterial abnormalities in patients with facial capillary haemangioma: presumed incomplete phenotypic expression of PHACES syndrome. Neuroradiology. 2001; 43: 934-940.
- Hess CP, Fullerton HJ, Metry DW, Drolet BA, Siegel DH, Auguste KI, et al. Cervical and intracranial arterial anomalies in 70 patients with PHACE syndrome. AJNR Am J Neuroradiol. 2010; 31: 1980-1986.
- Liao C, Fu F, Li R, Yang X, Xu Q, Li DZ. Prenatal diagnosis and molecular characterization of a novel locus for Dandy-Walker malformation on chromosome 7p21.3. Eur J Med Genet. 2012; 55: 472-475.
- Grinberg I, Northrup H, Ardinger H, Prasad C, Dobyns WB, Millen KJ. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet. 2004; 36: 1053-1055.
- Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009; 41: 1037-1042.
- Zanni G, Barresi S, Travaglini L, Bernardini L, Rizza T, Digilio MC, et al. FGF17, a gene involved in cerebellar development, is downregulated in a patient with Dandy-Walker malformation carrying a de novo 8p deletion. Neurogenetics. 2011; 12: 241-245.
- Darbro BW, Mahajan VB, Gakhar L, Skeie JM, Campbell E, Wu S, et al. Mutations in extracellular matrix genes NID1 and LAMC1 cause autosomal dominant Dandy-Walker malformation and occipital cephaloceles. Hum Mutat. 2013; 34: 1075-1079.
- Kumar R, Jain MK, Chhabra DK. Dandy-Walker syndrome: different modalities of treatment and outcome in 42 cases. Childs Nerv Syst. 2001; 17: 348-352.
- Alexiou GA, Sfakianos G, Prodromou N. Dandy-Walker malformation: analysis of 19 cases. J Child Neurol. 2010; 25: 188-191.
- Ecker JL, Shipp TD, Bromley B, Benacerraf B. The sonographic diagnosis of Dandy-Walker and Dandy-Walker variant: associated findings and outcomes. Prenat Diagn. 2000; 20: 328-332.
- Bolduc ME, Limperopoulos C. Neurodevelopmental outcomes in children with cerebellar malformations: a systematic review. Dev Med Child Neurol. 2009; 51: 256-267.
- Bolduc ME, Du Plessis AJ, Sullivan N, Khwaja OS, Zhang X, Barnes K, et al. Spectrum of neurodevelopmental disabilities in children with cerebellar malformations. Dev Med Child Neurol. 2011; 53: 409-416.
- Parisi MA, Dobyns WB. Human malformations of the midbrain and hindbrain: review and proposed classification scheme. Mol Genet Metab. 2003; 80: 36-53.
- Tortori-Donati P, Fondelli MP, Rossi A, Carini S. Cystic malformations of the posterior cranial fossa originating from a defect of the posterior membranous area. Mega cisterna magna and persisting Blake's pouch: two separate entities. Childs Nerv Syst. 1996; 12: 303-308.
- Nelson MD, Maher K, Gilles FH. A different approach to cysts of the posterior fossa. Pediatr Radiol. 2004; 34: 720-732.
- Cornips EM, Overvliet GM, Weber JW, Postma AA, Hoeberigs CM, Baldewijns MM, et al. The clinical spectrum of Blake's pouch cyst: report of six illustrative cases. Childs Nerv Syst. 2010; 26: 1057-1064.
- Ali ZS, Lang SS, Bakar D, Storm PB, Stein SC. Pediatric intracranial arachnoid cysts: comparative effectiveness of surgical treatment options. Childs Nerv Syst. 2014; 30: 461-469.
- Barkovich AJ, Kjos BO, Norman D, Edwards MS. Revised classification of posterior fossa cysts and cystlike malformations based on the results of multiplanar MR imaging. AJR Am J Roentgenol. 1989; 153: 1289-1300.
- Yildiz H, Yazici Z, Hakyemez B, Erdogan C, Parlak M. Evaluation of CSF flow patterns of posterior fossa cystic malformations using CSF flow MR imaging. Neuroradiology. 2006; 48: 595-605.
- Estroff JA, Scott MR, Benacerraf BR. Dandy-Walker variant: prenatal sonographic features and clinical outcome. Radiology. 1992; 185: 755-758.
- Boddaert N, Klein O, Ferguson N, Sonigo P, Parisot D, Hertz-Pannier L, et al. Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiology. 2003; 45: 320-324.
- Klein O, Pierre-Kahn A, Boddaert N, Parisot D, Brunelle F. Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv Syst. 2003; 19: 484-489.
- Kölble N, Wisser J, Kurmanavicius J, Bolthauser E, Stallmach T, Huch A, et al. Dandy-walker malformation: prenatal diagnosis and outcome. Prenat Diagn. 2000; 20: 318-327.
- Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet. 2009; 151: 326-340.
- Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013; 12: 894-905.
- Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013; 93: 915-925.
- Romani M, Micalizzi A, Kraoua I, Dotti MT, Cavallin M, Sztriha L, et al. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J Rare Dis. 2014; 9: 72.
- Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. 2014; 35: 137-146.
- Tuz K, Bachmann-Gagescu R, O'Day DR, Hua K, Isabella CR, Phelps IG, et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am J Hum Genet. 2014; 94: 62-72.
- Beck BB, Phillips JB, Bartram MP, Wegner J, Thoenes M, Pannes A, et al. Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum Mutat. 2014; 35: 1153-1162.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011; 364: 1533-1543.
- Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM, et al. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med. 2011; 17: 726-731.
- Aguilar A, Meunier A, Strehl L, Martinovic J, Bonniere M, Attie-Bitach T, et al. Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. Proc Natl Acad Sci U S A. 2012; 109: 16951-16956.
- Poretti A, Dietrich Alber F, Brancati F, Dallapiccola B, Valente EM, Boltshauser E. Normal cognitive functions in joubert syndrome. Neuropediatrics. 2009; 40: 287-290.
- Poretti A, Christen HJ, Elton LE, Baumgartner M, Korenke GC, Sukhudyan B, et al. Horizontal head titubation in infants with Joubert syndrome: a new finding. Dev Med Child Neurol. 2014; 56: 1016-1020.
- Poretti A, Vitiello G, Hennekam RC, Arrigoni F, Bertini E, Borgatti R, et al. Delineation and diagnostic criteria of Oral-Facial-Digital Syndrome type VI. Orphanet J Rare Dis. 2012; 7: 4.
- Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet. 2010; 47: 8-21.
- Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D, et al. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999; 14: 368-376.
- Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, et al. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004; 125: 125-134.
- Poretti A, Huisman TA, Scheer I, Boltshauser E. Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. AJNR Am J Neuroradiol. 2011; 32: 1459-1463.
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet. 2007; 15: 511-521.
- Dafinger C, Liebau MC, Elsayed SM, Hellenbroich Y, Boltshauser E, Korenke GC, et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J Clin Invest. 2011; 121: 2662-2667.
- Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet. 2011; 43: 601-606.
- Putoux A, Nampoothiri S, Laurent N, Cormier-Daire V, Beales PL, Schinzel A, et al. Novel KIF7 mutations extend the phenotypic spectrum of acrocallosal syndrome. J Med Genet. 2012; 49: 713-720.
- Lopez E, Thauvin-Robinet C, Reversade B, Khartoufi NE, Devisme L, Holder M, et al. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum Genet. 2014; 133: 367-377.
- Widjaja E, Blaser S, Raybaud C. Diffusion tensor imaging of midline posterior fossa malformations. Pediatr Radiol. 2006; 36: 510-517.
- Poretti A, Boltshauser E, Loenneker T, Valente EM, Brancati F, Il'yasov K, et al. Diffusion tensor imaging in Joubert syndrome. AJNR Am J Neuroradiol. 2007; 28: 1929-1933.
- Friede RL, Boltshauser E. Uncommon syndromes of cerebellar vermis aplasia. I: Joubert syndrome. Dev Med Child Neurol. 1978; 20: 758-763.
- Yachnis AT, Rorke LB. Neuropathology of Joubert syndrome. J Child Neurol. 1999; 14: 655-659.
- Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004; 36: 1008-1013.
- Engle EC. Human genetic disorders of axon guidance. Cold Spring Harb Perspect Biol. 2010; 2: a001784.
- Toelle SP, Yalcinkaya C, Kocer N, Deonna T, Overweg-Plandsoen WC, Bast T, et al. Rhombencephalosynapsis: clinical findings and neuroimaging in 9 children. Neuropediatrics. 2002; 33: 209-214.
- Ishak GE, Dempsey JC, Shaw DW, Tully H, Adam MP, Sanchez-Lara PA, et al. Rhombencephalosynapsis: a hindbrain malformation associated with incomplete separation of midbrain and forebrain, hydrocephalus and a broad spectrum of severity. Brain. 2012; 135: 1370-1386.
- Poretti A, Bartholdi D, Gobara S, Alber FD, Boltshauser E. Gomez-Lopez-Hernandez syndrome: an easily missed diagnosis. Eur J Med Genet. 2008; 51: 197-208.
- Sukhudyan B, Jaladyan V, Melikyan G, Schlump JU, Boltshauser E, Poretti A. Gómez-López-Hernández syndrome: reappraisal of the diagnostic criteria. Eur J Pediatr. 2010; 169: 1523-1528.
- Tully HM, Dempsey JC, Ishak GE, Adam MP, Curry CJ, Sanchez-Lara P, et al. Beyond Gómez-López-Hernández syndrome: recurring phenotypic themes in rhombencephalosynapsis. Am J Med Genet A. 2012; 158: 2393-2406.
- Poretti A, Alber FD, Bürki S, Toelle SP, Boltshauser E. Cognitive outcome in children with rhombencephalosynapsis. Eur J Paediatr Neurol. 2009; 13: 28-33.
- Tully HM, Dempsey JC, Ishak GE, Adam MP, Mink JW, Dobyns WB, et al. Persistent figure-eight and side-to-side head shaking is a marker for rhombencephalosynapsis. Mov Disord. 2013; 28: 2019-2023.
- Whitehead MT, Choudhri AF, Grimm J, Nelson MD. Rhombencephalosynapsis as a cause of aqueductal stenosis: an under-recognized association in hydrocephalic children. Pediatr Radiol. 2014; 44: 849-856.
- Barth PG, Majoie CB, Caan MW, Weterman MA, Kyllerman M, Smit LM, et al. Pontine tegmental cap dysplasia: a novel brain malformation with a defect in axonal guidance. Brain. 2007; 130: 2258-2266.
- Jissendi-Tchofo P, Doherty D, McGillivray G, Hevner R, Shaw D, Ishak G, et al. Pontine tegmental cap dysplasia: MR imaging and diffusion tensor imaging features of impaired axonal navigation. AJNR Am J Neuroradiol. 2009; 30: 113-119.
- Rauscher C, Poretti A, Neuhann TM, Forstner R, Hahn G, Koch J, et al. Pontine tegmental cap dysplasia: the severe end of the clinical spectrum. Neuropediatrics. 2009; 40: 43-46.
- Briguglio M, Pinelli L, Giordano L, Ferraris A, Germanò E, Micheletti S, et al. Pontine Tegmental Cap Dysplasia: developmental and cognitive outcome in three adolescent patients. Orphanet J Rare Dis. 2011; 6: 36.
- Rudaks LI, Patel S, Barnett CP. Novel clinical features in pontine tegmental cap dysplasia. Pediatr Neurol. 2012; 46: 393-396.
- Desai NK, Young L, Miranda MA, Kutz JW, Roland PS, Booth TN. Pontine tegmental cap dysplasia: the neurotologic perspective. Otolaryngol Head Neck Surg. 2011; 145: 992-998.
- Caan MW, Barth PG, Niermeijer JM, Majoie CB, Poll-The BT. Ectopic peripontine arcuate fibres, a novel finding in pontine tegmental cap dysplasia. Eur J Paediatr Neurol. 2014; 18: 434-438.
- Boltshauser E, Steinlin M, Martin E, Deonna T. Unilateral cerebellar aplasia. Neuropediatrics. 1996; 27: 50-53.
- Poretti A, Limperopoulos C, Roulet-Perez E, Wolf NI, Rauscher C, Prayer D, et al. Outcome of severe unilateral cerebellar hypoplasia. Dev Med Child Neurol. 2010; 52: 718-724.
- Merrill JD, Piecuch RE, Fell SC, Barkovich AJ, Goldstein RB. A new pattern of cerebellar hemorrhages in preterm infants. Pediatrics. 1998; 102: 62.
- Ranzini AC, Shen-Schwarz S, Guzman ER, Fisher AJ, White M, Vintzileos AM. Prenatal sonographic appearance of hemorrhagic cerebellar infarction. J Ultrasound Med. 1998; 17: 725-727.
- Yuksel A, Batukan C. Fetal cerebellar hemorrhage in a severely growth-restricted fetus: natural history and differential diagnosis from Dandy-Walker malformation. Ultrasound Obstet Gynecol. 2003; 22: 178-181.
- Ortiz JU, Ostermayer E, Fischer T, Kuschel B, Rudelius M, Schneider KT. Severe fetal cytomegalovirus infection associated with cerebellar hemorrhage. Ultrasound Obstet Gynecol. 2004; 23: 402-406.
- Lerner A, Gilboa Y, Gerad L, Malinger G, Kidron D, Achiron R. Sonographic detection of fetal cerebellar cavernous hemangioma with in-utero hemorrhage leading to cerebellar hemihypoplasia. Ultrasound Obstet Gynecol. 2006; 28: 968-971.
- Gorincour G, Rypens F, Lapierre C, Costa T, Audibert F, Robitaille Y. Fetal magnetic resonance imaging in the prenatal diagnosis of cerebellar hemorrhage. Ultrasound Obstet Gynecol. 2006; 27: 78-80.
- Malinger G, Zahalka N, Kidron D, Ben-Sira L, Lev D, Lerman-Sagie T. Fatal outcome following foetal cerebellar haemorrhage associated with placental thrombosis. Eur J Paediatr Neurol. 2006; 10: 93-96.
- Nomura ML, Barini R, de Andrade KC, Faro C, Marins M. Prenatal diagnosis of isolated fetal cerebellar hemorrhage associated with maternal septic shock. Prenat Diagn. 2009; 29: 169-171.
- Oza VS, Wang E, Berenstein A, Waner M, Lefton D, Wells J, et al. PHACES association: a neuroradiologic review of 17 patients. AJNR Am J Neuroradiol. 2008; 29: 807-813.
- Benbir G, Kara S, Yalcinkaya BC, Karhkaya G, Tuysuz B, Kocer N, et al. Unilateral cerebellar hypoplasia with different clinical features. Cerebellum. 2011; 10: 49-60.