
Case Report
Austin J Neurol Disord Epilepsy. 2017; 4(1): 1028.
A Rare Case of Unilateral Lower Limb Spasticity: Two Rare Mutations!
Mazumdar J¹*, Ghosh S², Mahato M², Chandra Ghosh K², Bhattacharya R² and Mondal G²
¹Department of Pediatrics, Calcutta National Medical College & Hospital, Calcutta, India
²Department of Neurology, Calcutta National Medical College & Hospital, Calcutta, India
*Corresponding author: Jayitri Mazumdar, Department of Pediatrics, Calcutta National Medical College & Hospital, Calcutta, India
Received: December 30, 2016; Accepted: January 19, 2017; Published: January 23, 2017
Abstract
The SPG11 gene was first analyzed, revealing homozygous or compound heterozygous mutations in 30/97 (30.9%) of pro bands, the largest SPG11 series reported to date, and by far the most common cause of complex spastic paraplegia Hereditary spastic paraplegia (HSP) is a group of clinically and genetically heterogeneous neurological disorders characterized by path physiologic hallmark of length-dependent distal axonal degeneration of the corticospinal tracts. HSP can be transmitted as an Autosomal Dominant (AD), Autosomal Recessive (AR), X-linked (XL), or mitochondrial manner. Here we describe a 15-yr-oldboy with abnormal posturing of right lower limb (dystonia and spasticity) during walking, who MRI is showed thinned out corpus callosum. Patients younger brother also had weakness in the lower limbs and Similar MRI findings. Mutation analysis revealed two rare variations in the SPG 11 gene not reported in medical literature till date.
Keywords: Hereditary spastic paraplegia (HSP); Autosomal recessive
Introduction
In its pure form, Hereditary spastic paraplegias (HSP) is usually transmitted as an autosomal trait; Symptoms usually begin in the third in the third or fourth decade, presenting as progressive spastic weakness beginning in the distal lower extremities. However, there are variants with relatively early onset mimicking cerebral palsy. HSP has a typically long survival as respiratory function is spared1. Late in the illness, there may be urinary urgency, incontinence and fecal incontinence. In pure forms of HSP, spastic leg weakness is often accompanied by posterior column abnormalities and bladder bowel dysfunction. By contrast, when recessively inherited HSP may have complex forms in which altered corticospinal and dorsal column function is accompanied by significant involvement of other regions of nervous system, including amyotrophy, mental retardation, optic atrophy and sensory neuropathy [1].
Here we present a 15 yr old boy with complex hereditary spastic paraparesis, which is asymmetric in involvement i.e. right side affected more than left, along with mild mental retardation and thin corpus callosum, showing two heterozygous variations in the SPG 11 gene at a time. These two variations are extremely rare and not reported in medical literature till date, giving a unique presentation in the index case with right lower limb dystonia with spasticity.
Case Presentation
15-yr-old-male, residing at Bangladesh was admitted in this hospital with chief complaints of insidious onset and gradually progressive weakness of both lower limbs for last 1 year. The weakness was distal to start with and gradually progressed to involve the proximal limb. There was abnormal posturing of the right foot especially during walking (Video 1) for the same duration which subsided during sleep.
Video
Please download Video here
Patient was alert and conscious with no alteration of sensorium. The illness was not preceded by fever, rash, joint pain. There was no associated gaze palsy, bulbar or other carnial nerve involvement. Patient had no bladder bowel abnormality, tingling numbness, self mutilating behaviour or any other abnormal movement except the dystonia of right foot as described before. Past history was uneventful. Family history reveals patients younger brother has similar complain.
On examination, higher mental function revealed a MMSE (mini mental score estimation) of 22/30 and IQ assessment showed mild mental retardation with an IQ score of 70. All the cranial nerves were normal in function. The right foot was inverted and plantiflexed during examination of gait. There were no appreciable weakness in the upper limbs, neck and axial muscles. Power was 5/5 in both the upper limbs and 4/5 in both lower limbs with planter bilateral extensor.
Laboratory investigations showed CBC, electrolytes within normal limit. CSF study showed nothing significant. Serum ceruloplasmin was 24mg/dl (within normal reference range) and slit lamp examination of eyes failed to demonstrate any K-F (Keisure- Fleshiers Ring). MRI revealed a thinned out corpus callosum (both in the index case and his younger brother) (Figure 1 - 2).
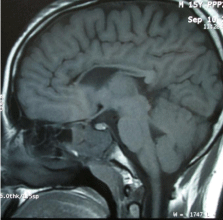
Figure 1: Sagital T2 W MRI showed thinned out corpus callosum in the index
case.
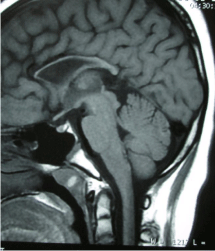
Figure 2: Sagital T2 W MRI showed thinned out corpus callosum in the
younger brother.
NCV (Nerve Conduction Velocity) of the lower limbs showed right seded tibial axonopathy. Keeping in mind the possibility of Hereditary Spastic Paraplegia (HSP) we went for mutation analysis of HSP gene panel. The result revealed two heterozygous variations in the SPG11 gene. A heterozygous single base pair insetion in exon 38 of the SPG11 gene (chr15:44858180dupA) that resulting in a frame shift and premature truncation of the protein 49 amino acids downstream to codon 2291 was detected. (This variant is not reported in both the 1000 genomes and EXAC databases.)
Another heterozygous missence variation in exon 21 of the SPG11 gene (chr15:44892728;G>G/A) that resulting in the amino acid substitution of Leucine for Proline at codon 1208 was also detected. (This P1208L variant is not present in the 1000 genomes databases and has a minor allele frequency of less than 0.001% in the EXAC database). The patient was discharged with baclofen, Vitamine E and methylecobalamine. Patient is still in follow up 6 monthly for a period of 1yr and had deteriorated further.
Discussion
There is an ever-expanding list of genes and genetic loci in the HSP family. Novel techniques such as exome sequencing are valuable in discovering new genes. Inheritance of most pure HSP is autosomal dominant, whereas complicated forms are more often autosomal recessive [2].
Spastic paraplegia 11 (SPG11) is an autosomal recessive complicated HSP (thin corpus callosum, neuropathy, cognitive impairment) due to mutations in the spatacsin gene on chromosome 15q13-q152. This protein is of unknown function and is predicted to contain a Glycosyl hydroxylase F1 signature (482-490 aa), a Leucine zipper (611-632 aa), a short coiled domain (1556-1590 aa), a Myb domain (1766-1774 aa), and 4 Transmembrane Domains (TM) located between amino acid residues 163-194, 200-240, 1239- 1267 and 1471-1493. Spatacsin is expressed most predominantly in cerebellum, cerebral cortex, hippocampus and pineal gland [3,4].
However, SPG11/KIAA1840 mutations have been also identified as a genetic cause of juvenile Amyotrophic Lateral Sclerosis type 5 (ALS5) in 40% of patients 4 and Kjellin syndrome (Orlén et al., 2009) characterized by central retinal degeneration, mental retardation and amyotrophy in addition to autosomal recessive HSP with thin corpus callosum. The SPG11 gene was first analyzed, revealing homozygous or compound heterozygous mutations in (30.9%) of probands, the largest SPG11 series reported till date, and by far the most common cause of complex spastic paraplegia with progressive clinical features and other neurological manifestations and magnetic resonance imaging defects [5].
On an average, patients become wheelchair-bound 16 years after disease onset. Cognitive impairment and thin corpus callosum are reliable phenotype predictors of SPG11; other common features are axonal polyneuropathy and cerebellar signs [6].
In our index case there is thinned out corpus callosum with spasticity in right lower limb to start with. Interestingly the younger brother presented with similar features like thinned out corpus callosum and mental retardation, though typical posturing of the lower limb was absent in the later as compared to index case. However, we could not proceed for the genetic analysis in the younger brother. Most confirmatory was the two genetic variations noted in the mutation analysis of theSPG11 gene of the index case as described above (Figure 3 showing mutations in sanger’s sequencing), not reported in medical literature till date , thus making the case really a rare one.
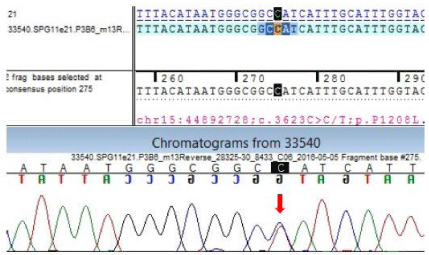
Figure 3: Sequence chromatogram and alignment to the reference sequence
showing the variation in exon 21 (chr15:44892728G>G/A; c.3623C>C/T;
p.P1208L) detected in the index case.
References
- Robert H. Brown, Jr. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disease. Harrison’s Principal of Internal Medicine. 2015; 19: 2634-2636.
- Fearon C, Murray B, Mitsumoto H. Disorders of Upper and Lower Motor Neurons. Bradley’s Neurology in Clinical Practice. 2016; 7: 1487-1488.
- Stevanin G, Santorelli FM, Azzedine H,Coutinho P, Chomilier Jacques, Denora PS, et al. Mutations in SPG11, encodingspatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007; 39: 366- 372.
- Orlacchio A. stem cells and neurological disease. 2010.
- Klebe S, Stevanin G, Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting Rev Neurol (Paris). 2015; 171: 505-530.
- Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms, Experimental Neurology. 2014; 261: 518–539.