
Special Issue - Parkinson’s Disease
Austin Neurol & Neurosci. 2017; 2(2): 1021.
BDNF in the Aged Brain: Translational Implications for Parkinson’s Disease
Mercado NM1, Collier TJ1,2, Sortwell CE1,2 and Steece-Collier K1,2*
¹Department of Translational Science & Molecular Medicine, College of Human Medicine, Michigan State University, USA
²Hauenstein Neuroscience Center, Mercy Health Saint Mary’s, USA
*Corresponding author: Steece-Collier K, Department of Translational Science & Molecular Medicine, Michigan State University, Grand Rapids, USA
Received: July 10, 2017; Accepted: September 12, 2017; Published: September 19, 2017
Abstract
Brain Derived Neurotrophic Factor (BDNF) is a member of the neurotrophin family of secreted growth factors. BDNF signaling is known to exert both chronic, pro-survival effects related to gene expression and protein synthesis (“canonical signaling”), and acute effects as a modulator of neurotransmission (“non-canonical signaling”). BDNF has received a great deal of attention for its role in neurodegenerative diseases including Huntington’s Disease (HD), Alzheimer’s Disease (AD), and Parkinson’s Disease (PD) and has been extensively reviewed elsewhere in this regard (e.g., [1-6]). However agingrelated changes in BDNF function and expression have been studied only rarely, with the majority of studies characterizing changes in structures such as the hippocampus and neocortex. In this review, we attempt to briefly summarize the extent of the existing literature on age-related BDNF changes, and discuss the relevance of these changes as a factor potentially impacting therapeutics in aged parkinsonian subjects.
Keywords: Brain derived neurotrophic factor; Parkinson’s disease; Aging; Therapeutics
Introduction
Neurotrophins are important regulators of neuronal survival, development, maintenance, and plasticity. The mammalian neurotrophin family consists of four proteins: Nerve Growth Factor (NGF), Brain-Derived Neurotrophic Factor (BDNF), Neurotrophin-3 (NT3), and Neurotrophin-4/5 (NT4/5) [7]. Of these, BDNF was the second to be discovered, after the groundbreaking discovery of NGF in the 1950s by Rita Levi-Montalcini, Stanley Cohen, and Viktor Hamburger, for which Levi-Montalcini and Cohen were awarded the Nobel Prize in Physiology or Medicine in 1986 ([8-13], but see [14] for review). In 1982, Yves-Alain Barde and colleagues isolated BDNF for the first time from pig brain [15]. In the 35 years since its discovery, BDNF has been intensively studied, as evidenced by the thousands of publications currently available on this particular neurotrophic factor.
Despite the plethora of existing BDNF literature, potential aging-induced changes in BDNF expression and signaling have been studied only sparingly. Here, we briefly discuss age-related changes in BDNF and its association with Parkinson’s disease (PD), a neurodegenerative disease whose primary risk factor is advanced age. We focus our discussion on putative age-related changes in BDNF-tyrosine receptor kinase B (trkB) signaling dynamics, protein/ messenger RNA (mRNA) expression within the striatum, and implications for therapeutic approaches for the treatment of PD.
BDNF: A Well-Studied Molecule
The BDNF gene: many transcripts, one protein
The rat BDNF gene was first described in 1993 by Timmusk et al. [16], whose findings have been expanded in more recent studies ([17-19], see [1] for detailed review). Briefly, the rodent BDNF gene contains eight untranslated 5’ exons (exons I-VIII), the majority of which are linked to separate, distinct promoters, and one proteincoding exon (exon IX) [19]. In humans, the BDNF gene is even more complex, with 11 distinct exons controlled by nine promoters [20]. Due to multiple promoters, alternative splicing, and polyadenylation, the various BDNF exons produce a large variety of BDNF transcripts, which control tissue-, development-, and stimulus-specific BDNF protein expression [16,19,21]. Remarkably, each transcript encodes the same BDNF protein [19,22]. Of relevance to neurodegenerative disease, the structure of human BDNF is closely related to rat and mouse, and all exons defined in humans are also expressed in mouse and rat, except for human exons VIIB and VIII [23].
Neurotrophin secretion and activation of downstream signaling pathways
All neurotrophins, including BDNF, are initially synthesized as pre-pro-neurotrophin precursors [24]. The pre-mRNA sequence, which gives rise to the signal peptide within the final protein product, directs newly-generated neurotrophins to ribosomes located in the endoplasmic reticulum (ER) where the pre-sequence is cleaved and the initial protein is translated. The resulting pro-neurotrophins accumulate in the trans-golgi network (TGN) and are sorted into secretory vesicles [24]. The pro-sequence is typically cleaved from the final protein product by protein convertases within the TGN or secretory vesicles [24], though uncleaved pro-neurotrophins are also released from cells and are known to activate the p75 neurotrophin receptor (p75NTR), initiating apoptosis [25,26]. However, following the discovery of the Vps10 sortilin family member SorCS2 (Sortilinrelated CNS expressed 2) as a co-receptor for p75NTR and trkB [27], it has become apparent that pro-neurotrophins acting via the p75NTR/ sortilin or p75NTR/SorCS2 receptor complex may also regulate signaling pathways that in turn regulate processes such as synaptic activity and pruning, and network reorganization [28]. Furthermore, pro-BDNF is thought to be a key regulator of neuronal circuitry and plasticity, especially in early postnatal periods, as it has been shown in the hippocampus to negatively regulate dendritic complexity, impair long-term potentiation, and enhance long-term depression in the hippocampus [29]. On the other hand, the mature BDNF protein (mBDNF) activates a member of the tyrosine receptor kinase family, trkB, through which it also impacts neuronal morphology and synaptic plasticity albeit through mechanisms distinct from pro- BDNF [29], as well as the pro-survival and protein synthesis signaling pathways typically associated with BDNF activity.
BDNF is released from presynaptic terminals via two distinct pathways mediated by separate populations of secretory vesicles. When pro-BDNF is cleaved in the TGN, the mature protein is sorted into small secretory granules of the constitutive secretion pathway, where it is transported to the cell membrane and released in a stimulusindependent manner [24]. However, when sorted into larger vesicles of the regulated secretion pathway, pro-BDNF is cleaved within the vesicles and the mature protein is released in a strictly calciumdependent manner [24]. Importantly, BDNF is almost exclusively sorted to and released from the regulated pathway. Indeed, Brigadski and colleagues showed that BDNF was preferentially targeted to the regulated pathway in 98% of cultured hippocampal neurons, and that fusing the usually constitutively-released NT-4 to the BDNF pre-pro-sequence more efficiently targeted NT-4 to the regulated pathway [30]. Regulated release of BDNF occurs following neuronal stimulation, for example, after high-frequency electrical stimulation or potassium-induced depolarization. This was demonstrated for the first time in early experiments using cultured hippocampal neurons [31,32] and confirmed in later studies (e.g., [33-37]). In addition, it is relevant to note that the activity-dependent regulation of BDNF transcription is controlled primarily through exons I and IV of the BDNF gene (human BDNF exon IV is equivalent to exon III in rat) [25,38].
Trk receptors are composed of intracellular tyrosine kinase domains and extracellular Immunoglobulin G (IgG) domains that bind ligands. Synaptically released BDNF dimerizes and binds to trkB, which initiates trkB dimerization and autophosphorylation of intracellular tyrosine residues [39]. Subsequently, three major intracellular signaling cascades are known to be activated: the Phospholipase Cγ (PLCγ), Phosphatidylinositol 3-Kinase (PI3K), and Extracellular signal-Regulated Kinase (ERK) pathways (see [39- 41] for detailed reviews). These pathways ultimately regulate gene expression and protein translation of their downstream targets. Furthermore, by activating these signaling cascades, BDNF also enhances synaptic transmission and membrane excitability. For example, at glutamatergic synapses such as those formed between cortical afferents to the striatum and striatal medium spiny neurons (MSNs), BDNF-trkB signaling stimulates an increase in the number of docked vesicles within active zones at synapses, and also alters activation kinetics of N-Methyl-D-Aspartate (NMDA) receptors and inhibitory Gamma-Amino Butyric Acid (GABA) receptors in the postsynaptic membrane (see [42] for review). BDNF also regulates actin dynamics and spine remodeling via activation of specific kinasemediated signaling cascades, including TIAM1/Rac1 and ROCK/ LIMK 1 pathways (e.g., [43,44]). On the other hand, pro-BDNF interaction with p75NTR activates three additional signaling pathways that result in the activation of NF-κB, Jun kinase, or RhoA, which in turn activate pro-survival genes, pro-apoptotic genes, or growth cone motility processes, respectively [41].
BDNF signaling is made more complex by co-receptor binding. For example, the binding of BDNF to trkB can be enhanced by trkB association with the p75NTR receptor [39], and p75NTR association with the sortilin receptors appears to mediate pro-apoptotic actions of p75NTR receptor activation [41]. Interestingly, the Vps10 sortilin family member SorCS2 was recently identified as an additional coreceptor for both trkB and p75NTR, and was shown to mediate BDNF-dependent plasticity, and bind to trkB in an activity-dependent manner [27]. It is clear that BDNF transcription, translation, secretion, and signaling are mediated via exquisitely complex mechanisms that produce a variety of different outcomes, from promoting cell apoptosis to controlling spine dynamics and synapse remodeling. This complexity and the divergent functional properties of BDNF present a unique opportunity for BDNF production and/ or signaling processes to become aberrant and accordingly have devastating consequences in association with its dysfunction, a topic of considerable interest in neurodegenerative disease.
BDNF and Parkinson’s Disease
Interest in a role for BDNF in PD stems from 1) its documented effects in promoting the survival and function of Substantia Nigra (SN) Dopamine (DA) neurons, and 2) its structural and functional influence on striatal MSNs, the principal target neurons of SN DA afferents. Indeed, more than 20 years of research demonstrates that BDNF is a critical factor for the viability of SN DA neurons. Specifically, BDNF supports the survival of SN neurons in vivo and is protective against multiple neurotoxin insults in vitro and in vivo [45- 49]. Further, haplo insufficiency of the BDNF receptor trkB in mice is associated with progressive degeneration of SN DA neurons, and in association with aging, results in excessive accumulation of alphasynuclein in remaining neurons [50]. Not only is BDNF important for nigral DA neuron survival, it is also important for the maturation and function of these neurons as evidenced by its ability to stimulate motor behavior, electrical activity of SN neurons, and DA turnover in the nigrostriatal system [46,48,51].
In the striatum BDNF supports survival of the immature MSNs, promotes maturation of these neurons, and facilitates establishment of striatal connections during brain development [52,53]. BDNF also is known to play an important role in dendritic spine dynamics and actin remodeling of MSNs in the adult brain, sculpting the structure and function of synapses (e.g., [54-57]). It also is implicated as a critical factor controlling dendritic spine density in many brain regions [40,46,58-60], the importance of which is detailed below. Striatal BDNF is primarily derived from cortical afferents that project to the striatum, but also from ascending SN DA afferents that release BDNF in an activity-dependent manner. Upon release, the interaction of BDNF with trkB receptors [40,44,61,62] located on striatal MSNs initiates signaling pathways involved in dendritic spine and synapse dynamics.
In PD, nigral DA neurons that send their afferent axons to the striatum degenerate, leading to detrimental structural changes in the striatum (e.g., decreased spine density, altered synaptic connections, etc., as described in [54-58]) and both motor and nonmotor symptoms. To date, there is strong evidence linking BDNF to PD pathology in the SN in individuals with PD. For instance, postmortem analysis of PD brains reveals a significant decrease in BDNF mRNA and protein in neurons of the SN pars compacta [59,60] and serum levels of BDNF correlate with the severity of motor symptoms [61], findings that could suggest a link between decreased BDNF and SN DA neuron death in PD. In addition, there is a compelling attribute of altered BDNF signaling that may pose a potential risk factor in the aged parkinsonian population in response to a variety of therapeutics. Specifically, a common Single Nucleotide Variant (SNV) in the BDNF gene that encodes the protein BDNF is present in the human population (rs6265; Val66Met) [62,63]. In this SNV, there is a Methionine (Met) substitution for Valine (Val) at codon 66 (Val66Met). The Met allele of the BDNF SNV rs6265 has a prevalence of 40.6% in the general population (Major/Minor or Val/Met = 35.4%, Minor/Minor or Met/Met = 5.2%, allelic frequency assuming Hardy-Weinberg) [64]. Both the heterozygous major allele (Val/ Met) and homozygous minor allele (Met/Met) of the BDNF SNV result in decreased activity-dependent release of BDNF by disruption of packaging into secretory vesicles, whereas constitutive levels of BDNF remain unaffected [65]. Although there are multiple SNVs in the BDNF gene, rs6265 is relatively unique in that this SNV is in the BDNF coding region, has a direct and well-studied impact on BDNF protein function, and is relatively prevalent within the human population. As discussed above, the majority of BDNF in the adult brain is released from neurons via the regulated secretory pathway; therefore, the impact of the BDNF SNV rs6265 leads to a significant decrease in available BDNF [65] in approximately 40% of the human population. While the rs6265 SNV does not appear to impact the clinical features of PD [66] and seems unlikely to play a major role in PD pathogenesis ([67-69]; however see [70]), it remains controversial whether subjects with this BDNF SNV risk allele are more susceptible to induction of levodopa-induced dyskinesias [71,72]. The full nature and extent of the influence of the rs6265 SNV on PD therapeutics, the potential decrease in BDNF signaling in aged parkinsonian brain, and/or the interaction of these factors remains uncertain. However, we contend that these factors warrant further investigation based on the discussion presented in the remainder of this review.
BDNF, Aging, and the Parkinsonian Striatum
Aging is the primary risk factor for PD, yet the impact of aging on therapeutic responses in PD, especially in preclinical studies, has received sparse attention (for review [73,74]). It is striking that what is known about the interaction of aging with PD and DA depletion strongly implicates aging as a significant factor in limitations of therapeutics in PD [73]. There are abundant data demonstrating that within the aged, DA depleted striatum, abundant pathology develops. For example, it is well documented in postmortem PD striatum that there are significant alterations of the most prominent neuron population, the MSNs [58,75,76], with not only a significant reduction in the length of their dendrites compared to age-matched controls, but also the remaining dendrites often show few to no spines [58].
Research over the past decade has begun to elucidate how and why these pathological changes in the MSNs of the parkinsonian striatum can impact PD therapeutics. Just briefly, the striatal MSNs have an extensive dendritic arbor that is studded with numerous dendritic spines. These spines are critical cytoarchitectural units that receive massive cortical glutamatergic motor information via afferent terminals that synapse onto the heads of the spines. The SN DA afferent terminals make synaptic contact onto the necks of the same spines as the cortical afferents, serving to modulate cortical input to the MSNs (for review [77-79]). Multiple studies demonstrate that loss of dendritic spines on striatal MSNs associated with DA depletion is associated with induction of levodopa and graft-induced dyskinesias (for review see [80]). Further, inferior behavioral recovery in aged parkinsonian rats is associated with evidence of inferior synaptic integration between graft and host [81], and preserving dendritic spine density on striatal MSNs in the parkinsonian striatum significantly improves therapeutic response to DA neuron replacement strategies [78].
As discussed above, BDNF is a critical factor controlling dendritic spine density [40,46,58-60] and sculpting the structure and function of synapses (e.g., [56,57]). As such, an environment of compromised BDNF signaling would be expected to negatively impact the integrity and/or function of these critical striatal cytoarchitectural elements, impact basal ganglia function, and accordingly impact responsiveness of individuals to therapeutic interventions. What is known about BDNF signaling in the aged, parkinsonian striatum? Existing evidence generally suggests that there is no change in BDNF mRNA or protein expression in brain across the lifespan; however, results appear to differ depending on the brain structure and the rat strain studied (see Table 1) [82]. In 1993, Lapchak and colleagues were the first to examine changes in trkB and BDNF mRNA expression in the aging rat brain [83]. Using northern blot and in situ hybridization techniques, Lapchak et al. [83] showed that the prevalence and regional distribution of BDNF and trkB mRNA within the hippocampus did not change with age. Similarly, Narisawa-Saito et al. [84] showed several years later that BDNF protein expression did not change with age in the hippocampus and frontal cortex of male Fischer 344 (F344) rats, though BDNF mRNA was significantly increased in the aged hippocampus. Croll and colleagues [85] later examined trkB and BDNF expression in various regions of the aging brain of male Sprague Dawley rats and of the multiple structures examined, BDNF protein was significantly decreased only in the midbrain of aged rats compared to young rats, and BDNF mRNA was significantly decreased only in the pons. Additional studies in both rats and human postmortem tissue identified various different, sometimes contradictory, age-related changes in trkB and BDNF expression with age (e.g., [86]). Thus, while there appears to be a the lack of consensus on age-related change in BDNF expression, which appears to vary between brain regions, an age-related decrease in trkB expression has been more consistently reported regardless of the structure examined (Table 1).
![]()
Publication
Subjects
Region Studied
TrkB studied?
BDNF studied?
Change with Age
Lapchak et al. (1993)
Male SD & F344 rats
Hippocampus
mRNA
mRNA
No change
Narisawa –Saito et al. (1996)
Male F344 rats
Hippocampus Frontal Cortex
Protein/ mRNA
�BDNF mRNA in hippocampus
No change in BDNF protein in either structure
Croll et al. (1998)
Male SD rats
various regions throughout brain, including striatum
mRNA
Protein/ mRNA
¯BDNF protein in midbrain only
¯BDNF mRNA in pons only
¯trkB Mrna in various regions
Romankzyk et al. (2002)
Postmortem human tissue
Prefrontal Cortex
mRNA
¯ full-length trkB mRNA with aging in all cortical layers
Webster et al.(2002)
Postmortem human tissue
Prefrontal Cortex
mRNA
No change
Silhol et al. (2005)
Male SD rats
Hippocampus Hypothalamus
Protein/ mRNA
Protein/ mRNA
¯ full-length trkB protein, no change in trkB Mrna
No change in BDNF protein or mRNA in either structure
Webster et al. (2006)
Postmortem human tissue
Hippocampus Temporal Cortex
mRNA
mRNA
¯ trkB RNA in both structures
¯ BDNF mRNA in cortex
No change in BDNF mRNA in hippocampus
Rage et al. (2007)
Male SD rats
Pitutary
Protein/ mRNA
Protein/ mRNA
¯ trkB protein
� BDNF mRNA
No change in BDNF protein or trkB mRNA
Chapman et al. (2012)
F344/Brown Norway hybrid rats
Hippocampus
mRNA
¯ BDNF mRNA in CA1 and CA3
No change in dentate gyrus
Calabrese et al. (2013)
Male Wistar Han rats
Hippocampus Prefrontal Cortex
Protein
Protein/ mRNA
¯ BDNF mRNA in both structures
¯ BDNF protein in both structures
¯ trkB protein in both structures
Perovic et al. (2013)
Wistar rats
Hippocampus Cortex
Protein
Protein/ mRNA
� BDNF mRNA in cortex
¯ BDNF mRNA in hippocampus
¯ full-length trkB protein in both structures
No change in BDNF protein
Tong et al. (2015)
Adult cats
Lateral Geniculate Nucleus
Protein
Protein
¯ BDNF and trkB proteins
Table 1: Summary of studies examining age-related changes in BDNF and trkB expression.
Returning to the question of whether there is a change of BDNF signaling in the aged striatum that could impact individuals with PD, it is noteworthy that in our review of the literature only a single study by Croll and colleagues [85] has assessed changes in BDNF and trkB expression within the aged striatum, where no significant changes were found. Although current evidence generally supports the idea that BDNF expression in the brain does not change with age (e.g., [85], but see Table 1 for additional details), studies have shown impaired ability of the aged brain to produce BDNF in response to stress [87,88] and reduced expression of the regulators of BDNF transcriptional activation including neuronal PAS domain protein 4 (Npas4) and cyclic Adenosine Mono Phosphate (cAMP) responsive element-binding protein (Creb) compared to the young brain [86]. Taken together, these data suggest a diminished capacity of the aged brain to transcribe, release, and/or respond to BDNF. Continued systematic investigations are warranted to fully understand the impact of aging in BDNF signaling, and its implication for therapeutics for multiple neurodegenerative conditions.
Implications of Dysfunctional BDNF for Therapeutics in PD
Within the striatum two main subsets of MSNs exist: those of the direct pathway (dMSNs) and those of the indirect pathway (iMSNs). Though morphologically similar, these two cell populations express distinct receptors and proteins (e.g., dMSNs express D1 DA receptors and iMSNs express D2 DA receptors), and have different projection pathways and physiological functions (e.g., [89]). They also appear to be differentially affected by DA depletion and levodopa treatment in PD (reviewed in [80]). In addition, data from a recent study by Gagnon and colleagues [90] indicate that a smaller population of MSNs exhibiting both D1 and D2 DA receptors (about 1.9% in the dorsal striatum) within the mouse striatum are morphologically distinct, and are impacted by DA depletion in a manner that is distinct from D1 MSNs or D2 MSNs. Interestingly, previous studies have demonstrated that in the nucleus accumbens (NAc) [91], in tissue containing both NAc and striatum used for fluorescence activated cell sorting [92], and in striatum during development [53], the majority of trkB mRNA and protein is in iMSNs. Further, when trkB is selectively deleted in striatal MSNs, only iMSNs are adversely affected, as indicated by a 63-80% loss of these neurons following trkB deletion, while dMSNs remain unaffected [53]. Also, iMSNs have been shown to degenerate first in Huntington’s disease (HD), and are more affected by BDNF down regulation in a heterozygous BDNF transgenic mouse model [93,94]. The work of Baydyuk et al. [52,53] suggests a causative role for differential trkB expression in MSN subtypes in vulnerability to development and disease-associated decline in MSN survival. Recent data from our laboratory support the hypothesis that overall striatal trkB mRNA expression measured using in situ hybridization declines with advanced age, and reveal for the first time that this age-related effect is restricted to iMSNs (Figure 1). Of note, this is the population of MSNs that also shows loss of dendritic spines in response to DA depletion ([54]; reviewed in [80]). The importance of this phenomenon for the pathophysiology of PD remains to be elucidated. Additional evidence from our laboratory demonstrates that dendritic spine density on striatal MSNs declines in rats in association with advanced age (Figure 2).
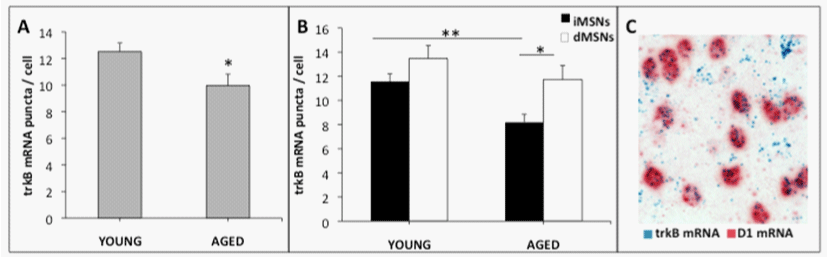
Figure 1: Dual-label RNAscope® in situ hybridization shows a selective loss of trkB mRNA in D2 receptor containing iMSNs in the aged striatum. A) Total trkB
mRNA expression is significantly reduced in MSNs of the dorsolateral striatum in aged compared to young Fischer 344 rats. B) TrkB mRNA decreased significantly
with age in indirect pathway MSNs containing D2 receptor mRNA, but not direct pathway MSNs containing D1 receptor mRNA. C) Representative 60x brightfield
microscopy image of dual-labeled striatal D1 DA receptor and trkB mRNA. Experimental Details: Young (4 m.o.) and aged (20 m.o.) F344 rats were perfused with
heparinized saline followed by 1.5% paraformaldehyde. Brains were post-fixed in 4% paraformaldehyde and cryoprotected before sectioning on a microtome. 20-
μm sections were dual-labeled for trkB and either D1 or D2 dopamine receptor RNA with the RNAscope® 2.5 Duplex manual assay for in situ hybridization. Images
of the dorsolateral striatum were taken at 60x magnification and the number of blue trkB mRNA puncta per cell was manually quantified using ImageJ software.
Four images per animal were used for analysis of each dopamine receptor probe.
*p < 0.05, **p < 0.01; Young N = 5, Aged N = 5.
Similar to the sparse data on the impact of age on striatal BDNF signaling, there is to the best of our knowledge but a single study examining the impact of advanced age on striatal dendritic spine density. This is notable given the abundance of data demonstrating that neurons within the hippocampus, cortex, and NAc show agingrelated loss of dendritic spines [95-99]. In the single study examining the impact of advanced age on dendritic spine density in the striatum, Levine and colleagues [100] directly compared changes in striatal MSN dendritic spine density between young and aged cats. They observed a 49% reduction in striatal dendritic spine density of aged (15-18 years old) cats compared to young (1-3 years old) cats. Our recent data (Figure 2) corroborate a loss of dendritic spines in the aged striatum of rats. Further, while spine loss related to DA depletion can be prevented pharmacologically with CaV1.3 calcium channel antagonists [54,78], our data demonstrate for the first time that agingrelated spine loss is NOT compensated for through this mechanism (Figure 2). Due to our observation that trkB mRNA was significantly decreased only in iMSNs of aged rats (Figure 1) and further evidence indicating that MSN spine density is decreased in BDNF knock-out animals [101-103], it is reasonable to suggest that age-related loss of spine density would be more significantly reduced in iMSNs than dMSNs. Considering that iMSNs and dMSNs have been shown to respond differently to DA depletion and levodopa treatment in animal models of PD (e.g., [104,105] but also see [80]), there is reason to infer that differential age-related changes in trkB expression may contribute to this phenomenon. A thorough characterization of this phenomenon is needed in the important preclinical rat model to understand 1) which MSN population is affected in the aged striatum, 2) whether a decrease in BDNF signaling, specifically the age-related decrease in trkB, underlies this age-related striatal spine loss, and 3) what the consequences of striatal synaptopathology secondary to dendritic spine loss are in clinical medicine.
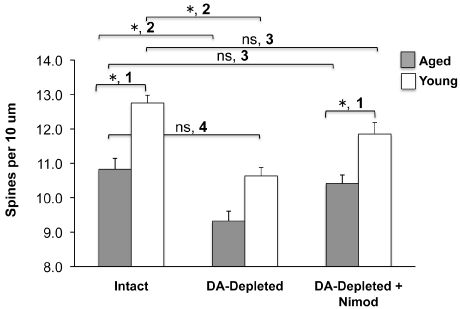
Figure 2: Dendritic spine dynamics in young and aged rats. ‘1’: Young rats
(3mo) have significantly more spines compared to aged rats (20mo). ‘2, 3’:
DA-depletion related spine loss (‘2’) is reversible with CaV1.3 calcium channel
antagonism (‘3’). ‘4’: The aged “normal” striatum has the same density of
dendritic spines as the young parkinsonian striatum. Experimental Details:
Young and aged male Sprague Dawley rats were lesioned with 6OHDA. One
cohort from each age group received vehicle pellets and a second received
slow-release subcutaneous pellets containing the CaV1.3 calcium channel
inhibitor nimodipine. Pellets were implanted 10 days after lesion, a time when
striatal MSNs show significant dendritic spine loss. Rats were sacrificed 3wks
after pellet implantation for spine density analysis with Golgi impregnation
techniques. MSNs were first visualized at low-power magnification and
selected for reconstruction based on the quality of Golgi–Cox impregnation.
Neurons selected for analysis were required to have at least four primary
dendrites that radiated from the soma in an arc encompassing 360° and to
not have excessive dendritic overlap with neighboring cells. Spine density
was quantified on three to six dendrites in N=3 young intact, N=3 DA-depleted
and N=4 DA-depleted+Nimod rats, and or 6, 5, or 8 N=6 aged intact, N=5 DAdepleted
and N=8 DA-depleted+Nimod rats.
* = significant difference p<0.05; ns = not significant; one-way ANOVA with
post hoc Tukey’s multiple comparisons test. Nimod = Nimodipine
Summary
In PD, for which the primary risk factor is aging, there is progressive loss of dendritic spines on MSNs. Dopamine depletion has a wellestablished link to striatal spine retraction (e.g., [54]; reviewed in [78]). However, data from our lab and Levine et al. [100] demonstrate that there also is loss of striatal spine density in association with advanced age, and our data reveal that the yet unknown mechanism is independent of CaV1.3 calcium channel activity. There is strong rationale to suggest that impaired BDNF signaling exists in the aged striatum, and that this may be further exacerbated in individuals with the rs6265 BDNF SNV. The critical nature of BDNF signaling in the nigrostriatal system, particularly its role in maintaining dendritic spine density and synapse function, would predict that impaired BDNF signaling in association with aging and/or the rs6265 SNV would significantly and negatively impact plasticity and function of the basal ganglia in health and disease. As discussed above, the loss of dendritic spines is associated with waning efficacy of levodopa and dyskinesia side-effect development, as well as abnormal “re-wiring” in experimental regenerative approaches of grafting of embryonic DA neurons into the parkinsonian striatum [58,77-79,81,106]. Despite the practical issues associated with aging animals in preclinical research, there remains a great need for exploration of the impact of aging on BDNF signaling in the nigrostriatal system and its impact on mainstream pharmacotherapies as well as experimental regenerative therapies in PD that continue to be of significant interest in the field. Continued investigations will be critical to the development of more efficacious therapeutics for the treatment of aging-related neurodegenerative diseases affecting the striatum, including PD.
References
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol. 2007; 81: 294-330.
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009; 5: 311-322.
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013; 14: 401416.
- Baydyuk M, Xu B. BDNF in Huntington’s Disease: Role in Pathogenesis and Treatment. Huntington’s Disease - Core Concepts and Current Advances. 2012.
- Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol. 2001; 63: 71-124.
- Binder DK, Scharfman HE. Brain-derived Neurotrophic Factor. Growth Factors. 2004; 22: 123-231.
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001; 24: 677-736.
- Levi-Montalcini R, Hamburger V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J Exp Zool. 1951; 116: 321-361.
- Levi-Montalcini R, Hamburger V. A diffusible agent of mouse sarcoma, producing hyperplasia of sympathetic ganglia and hyperneurotization of viscera in the chick embryo. J Exp Zool. 1953; 123: 233-287.
- Cohen S, Levi-Montalcini R, Hamburger V. A nerve growth-stimulating factor isolated from sarcomas 37 and 180. Proc Natl Acad Sci USA. 1954; 40: 1014-1018.
- Cohen S, Levi-Montalcini R. A nerve growth-stimulating factor isolated from snake venom. Proc Natl Acad Sci USA. 1956; 42: 571-574.
- Levi-Montalcini R, Cohen S. In vitro and in vivo effects of a nerve growthstimulating agent isolated from snake venom. Proc Natl Acad Sci USA. 1956; 42: 695-699.
- Cohen S. Purification of a nerve-growth promoting protein from the mouse salivary gland and its neuro-cytotoxic antiserum. Proc Natl Acad Sci USA. 1960; 46: 302-311.
- Levi-Montalcini R. The Nerve Growth Factor: Thirty-Five Years Later. Biosci Rep. 1987; 7: 681699.
- Barde YA, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982; 1: 549-553.
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, et al. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993; 10: 475-489.
- Liu QR, Walther D, Drgon T, Polesskaya O, Lesnick TG, Strain KJ, et al. Human Brain Derived Neurotrophic Factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2005; 134: 93-103.
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006; 1067: 1-12.
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007; 85: 525-535.
- Pruunsild P, Kazantseval A, Aid T, Palm K, Timmusk T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics. 2007; 90: 397-406.
- Timmusk T, Belluardo N, Persson H, Metsis M. Developmental regulation of brain-derived neurotrophic factor messenger RNAs transcribed from different promoters in the rat brain. Neuroscience. 1994; 60: 287-291.
- Greenberg ME, Xu B, Lu B, Hempstead BL. New Insights in the Biology of BDNF synthesis and release: Implications in CNS function. J Neurosci. 2009; 29: 12764-12767.
- Bathina S, Das UN. Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci. 2015; 11: 1164-1178.
- Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Prog Neurobiol. 2003; 69: 341-374.
- Hempstead BL. Brain-Derived Neurotrophic Factor: Three Ligands, Many Actions. Trans Am Clin Climatol Assoc. 2015; 126: 9-19.
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005; 25: 5455-5463.
- Glerup S, Bolcho U, Mølgaard S, Bøggild S, Vaegter CB, Smith AH, et al. SorCS2 is required for BDNF-dependent plasticity in the hippocampus. Mol Psychiatry. 2016; 21: 1740-1751.
- Gibon J, Barker PA. Neurotrophins and Proneurotrophins. Neuroscientist. 2017.
- Yang J, Harte-Hargrove LC, Siao CJ, Marinic T, Clarke R, Ma Q, et al. ProBDNF negatively regulates neuronal remodeling, synaptic ransmission, and synaptic plasticity in hippocampus. Cell Rep. 2014; 7: 796-806.
- Brigadski T, Hartmann M, Lessmann V. Differential vesicular targeting and time course of synaptic secretion of the mammalian neurotrophins. J Neurosci. 2005; 25: 7601-7614.
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990; 9: 3545-3550.
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992; 9: 1081- 1088.
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brainderived neurotrophic factor from primary sensory neurons detected by ELISA in Situ. J Neurosci. 2000; 20: 7417-7423.
- Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002; 22: 10399-10407.
- Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, et al. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001; 21: 4469- 4477.
- Blochl A, Thoenen H. Characterization of Nerve Growth Factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995; 7: 1220-1228.
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, et al. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996; 7: 222-238.
- West AE, Pruunsild P, Timmusk T. Neurotrophins: transcription and translation. Handb Exp Pharmacol. 2014; 220: 67-100.
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003; 4: 299-309.
- Segal RA. Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci. 2003; 26: 299-330.
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006; 361: 1545-1564.
- Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. 2010; 3: 1.
- Lai KO, Ip NY. Structural plasticity of dendritic spines: the underlying mechanisms and its dysregulation in brain disorders. Biochim Biophys Acta. 2013; 1832: 2257-2263.
- Panja D, Bramham CR. BDNF mechanisms in late LTP formation: A synthesis and breakdown. Neuropharmacology. 2014; 76: 664-676.
- Collier TJ, Sortwell CE. Therapeutic potential of nerve growth factors in Parkinson’s disease. Drugs Aging. 1999; 14: 261-287.
- Altar CA, Boylan CB, Jackson C, Hershensont S, Miller J, Wiegand SJ, et al. Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc Natl Acad Sci USA. 1992; 89: 11347-11351.
- Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, et al. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. 1991; 350: 230-232.
- Sauer H, Fischer W, Nikkhah G, Wiegand SJ, Brundin P, Lindsay RM, et al. Brain-derived neurotrophic factor enhances function rather than survival of intrastriatal dopamine cell-rich grafts. Brain Res. 1993; 626: 37-44.
- Zhou J, Bradford HF, Stern GM. The response of human and rat fetal ventral mesencephalon in culture to the brain-derived neurotrophic factor treatment. Brain Res. 1994; 656: 147-156.
- von Bohlen und Halbach O, Minichiello L, Unsicker K. Haploinsufficiency for trkB and trkC receptors induces cell loss and accumulation of alphasynuclein in the substantia nigra. FASEB J. 2005; 19: 1740-1742.
- Altar CA, Boylan CB, Fritsche M, Jones BE, Jackson C, Wiegand SJ, et al. Efficacy of brain-derived neurotrophic factor and neurotrophin-3 on neurochemical and behavioral deficits associated with partial nigrostriatal dopamine lesions. J Neurochem. 1994; 63: 1021-1032.
- Altar CA, Boylan CB, Fritsche M, Jones BE, Jackson C, Wiegand SJ, et al. Efficacy of brain-derived neurotrophic factor and neurotrophin-3 on neurochemical and behavioral deficits associated with partial nigrostriatal dopamine lesions. J Neurochem. 1994; 63: 1021-1032.
- Baydyuk M, Xu B. BDNF signaling and survival of striatal neurons. Front Cell Neurosci. 2014; 8: 254.
- Baydyuk M, Russell T, Liao GY, Zang K, An JJ, Reichardt LF, et al. TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc Natl Acad Sci U S A. 2011; 108: 1669-1674.
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006; 9: 251-259.
- Ingham CA, Hood SH, Arbuthnott GW. Spine density on neostriatal neurones changes with 6-hydroxydopamine lesions and with age. Brain Res. 1989; 503: 334-338.
- Ingham CA, Hood SH, Taggart P, Arbuthnott GW. Plasticity of synapses in the rat neostriatum after unilateral lesion of the nigrostriatal dopaminergic pathway. J Neurosci. 1998; 18: 4732-4743.
- Ingham CA, Hood SH, van Maldegem B, Weenink A, Arbuthnott GW. Morphological changes in the rat neostriatum after unilateral 6-hydroxydopamine injections into the nigrostriatal pathway. Exp Brain Res. 1993; 93: 17-27.
- McNeill TH, Brown SA, Rafols JA, Shoulson I. Atrophy of medium spiny I striatal dendrites in advanced Parkinson’s disease. Brain Res. 1988; 455: 148-152.
- Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, et al. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol. 2000; 166: 127-135.
- Parain K, Murer MG, Yan Q, Faucheux B, Agid Y, Hirsch E, et al. Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport. 1999; 10: 557-561.
- Scalzo P, Kummer A, Bretas TL, Cardoso F, Teixeira AL. Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson’s disease. J Neurol. 2010; 257: 540-545.
- Bath KG, Lee FS. Variant BDNF (Val66Met) impact on brain structure and function. Cogn Affect Behav Neurosci. 2006; 6: 79-85.
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003; 112: 257-269.
- https://www.ncbi.nlm.nih.gov/SNP/sn.
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006; 314: 140-143.
- Svetel M, Pekmezovic T, Markovic V, Novakovic I, Dobricic V, Djuric G, et al. No association between brain-derived neurotrophic factor G196A polymorphism and clinical features of Parkinson’s disease. Eur Neurol. 2013; 70: 257-262.
- Hong CJ, Liu HC, Liu TY, Lin CH, Cheng CY, Tsai SJ. Brain-Derived Neurotrophic Factor (BDNF) Val66Met polymorphisms in Parkinson’s disease and age of onset. Neurosci Lett. 2003; 353: 75-77.
- Håkansson A, Melke J, Westberg L, Shahabi H, Buervenich S, Carmine A, et al. Lack of association between the BDNF Val66Met polymorphism and Parkinson’s disease in a Swedish population. Ann Neurol. 2003; 53: 823.
- Karakasis C, Kalinderi K, Katsarou Z, Fidani L, Bostantjopoulou S. Association of Brain-Derived Neurotrophic Factor (BDNF) Val66Met polymorphism with Parkinson’s disease in a Greek population. J Clin Neurosci. 2011; 18: 1744-1745.
- Karamohamed S, Latourelle JC, Racette BA, Perlmutter JS, Wooten GF, Lew M, et al. BDNF genetic variants are associated with onset age of familial Parkinson disease: GenePD Study. Neurology. 2005; 65: 1823-1825.
- Foltynie T, Cheeran B, Williams-Gray CH, Edwards MJ, Schneider SA, Weinberger D, et al. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2009; 80: 141-144.
- Kaplan N, Vituri A, Korczyn AD, Cohen OS, Inzelberg R, Yahalom G, et al. Sequence variants in SLC6A3, DRD2, and BDNF genes and time to levodopa-induced dyskinesias in Parkinson’s disease. J Mol Neurosci. 2014; 53: 183-188.
- Mercado NM, Collier TJ, Freeman T, Steece-Collier K. Repairing the Aged Parkinsonian Striatum: Lessons from the Lab and Clinic. J Clin Cell Immunol. 2016; 7: 1-7.
- Polinski NK, Manfredsson FP, Benskey MJ, Fischer DL, Kemp CJ, Steece- Collier K, et al. Impact of age and vector construct on striatal and nigral transgene expression. Mol Ther Methods Clin Dev. 2016; 3: 16082.
- Zaja-Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, et al. Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology. 2005; 64: 545-547.
- Stephens B, Mueller AJ, Shering AF, Hood SH, Taggart P, Arbuthnott GW, et al. Evidence of a breakdown of corticostriatal connections in Parkinson’s disease. Neuroscience. 2005; 132: 741-754.
- Soderstrom KE, Meredith G, Freeman TB, McGuire SO, Collier TJ, Sortwell CE, et al. The Synaptic Impact of the Host Immune Response in a Parkinsonian Allograft Rat Model: Influence on Graft-Derived Aberrant Behaviors. Neurobiol Dis. 2008; 32: 229-242.
- Soderstrom KE, O’Malley JA, Levine ND, Sortwell CE, Collier TJ, Steece- Collier K. Impact of dendritic spine preservation in medium spiny neurons on dopamine graft efficacy and the expression of dyskinesias in parkinsonian rats. Eur J Neurosci. 2010; 31: 478-490.
- Zhang Y, Meredith GE, Mendoza-Elias N, Rademacher DJ, Tseng KY, Steece-Collier K. Aberrant restoration of spines and their synapses in L-DOPA-induced dyskinesia: involvement of corticostriatal but not thalamostriatal synapses. J Neurosci. 2013; 33: 11655-11667.
- Bastide MF, Meissner WG, Picconi B, Fasano S, Fernagut PO, Feyder M, et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinsons disease. Prog Neurobiol. 2015; 132: 96-168.
- Collier TJ, O’Malley J, Rademacher DJ, Stancati JA, Sisson KA, Sortwell CE, et al. Interrogating the aged striatum: robust survival of grafted dopamine neurons in aging rats produces inferior behavioral recovery and evidence of impaired integration. Neurobiol Dis. 2015; 77: 191-203.
- Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008; 59: 201-20.
- Lapchak PA, Araujo DM, Beck KD, Finch CE, Johnson SA, Hefti F. BDNF and trkB mRNA expression in the hippocampal formation of aging rats. Neurobiol Aging. 1993; 14: 121-126.
- Narisawa-Saito M, Nawa H. Differential regulation of hippocampal neurotrophins during aging in rats. J Neurochem. 1996; 67: 1124-1131.
- Croll SD, Ip NY, Lindsay RM, Wiegand SJ. Expression of BDNF and trkB as a function of age and cognitive performance. Brain Res. 1998; 812: 200-208.
- Calabrese F, Guidotti G, Racagni G, Riva MA. Reduced neuroplasticity in aged rats: a role for the neurotrophin brain-derived neurotrophic factor. Neurobiol Aging. 2013; 34: 2768-2776.
- Yurek DM, Fletcher-Turner A. Differential expression of GDNF, BDNF, and NT-3 in the aging nigrostriatal system following a neurotoxic lesion. Brain Res. 2001; 891: 228-235.
- Collier TJ, Dung Ling ZL, Carvey PM, Fletcher-Turner A, Yurek DM, Sladek JR, et al. Striatal Trophic Factor Activity in Aging Monkeys with Unilateral MPTP-induced Parkinsonism. Exp Neurol. 2005.
- Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci. 2014; 17: 1022-1030.
- Gagnon D, Petryszyn S, Sanchez MG, Bories C, Beaulieu JM, De Koninck Y, et al. Striatal Neurons Expressing D1 and D2 Receptors are Morphologically Distinct and Differently Affected by Dopamine Denervation in Mice. Sci Rep. 2017; 7: 41432.
- Koo JW, Lobo MK, Chaudhury D, Labonte B, Friedman A, Heller E, et al. Loss of BDNF signaling in D1R-expressing NAc neurons enhances morphine reward by reducing GABA inhibition. Neuropsychopharmacology. 2014; 39: 2646-2653.
- Lobo MK, Covington HE, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010; 330: 385-390.
- Richfield EK, Maguire-Zeiss KA, Vonkeman HE, Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington’s disease patients. Ann Neurol. 1995; 38: 852-861.
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, et al. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J Neurosci. 2004; 24: 7727-7739.
- Duan H, Wearne SL, Rocher AB, Macedo A, Morrison JH, Hof PR. Agerelated dendritic and spine changes in corticocortically projecting neurons in macaque monkeys. Cereb Cortex. 2003; 13: 950-961.
- Feldman ML, Dowd C. Loss of dendritic spines in aging cerebral cortex. Anat Embryol. 1975; 148: 279-301.
- Uemura E. Age-related changes in the subiculum of Macaca mulatta: synaptic density. Exp Neurol. 1985; 87: 403-411.
- Von Bohlen Und Halbach O, Zacher C, Gass P, Unsicker K. Age-related alterations in hippocampal spines and deficiencies in spatial memory in mice. J Neurosci Res. 2006; 83: 525-531.
- Li M, Dai FR, Du XP, Yang QD, Zhang X, Chen Y. Infusion of BDNF into the nucleus accumbens of aged rats improves cognition and structural synaptic plasticity through PI3K-ILK-Akt signaling. Behav Brain Res. 2012; 231: 146- 153.
- Levine MS, Fisher RS, Hull CD, Buchwald NA. Postnatal development of identified medium-sized caudate spiny neurons in the cat. Brain Res. 1986; 389: 47-62.
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brainderived neurotrophic factor. J Neurosci. 2004; 24: 4250-4258.
- Saylor AJ, Meredith GE, Vercillo MS, Zahm DS, McGinty JF. BDNF heterozygous mice demonstrate age-related changes in striatal and nigral gene expression. Exp Neurol. 2006; 199: 362-372.
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010; 30: 1739-1749.
- Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Commun. 2014; 5: 5316.
- Suárez LM, Solís O, Caramés JM, Taravini IR, Solís JM, Murer MG, et al. L-DOPA treatment selectively restores spine density in dopamine receptor d2-expressing projection neurons in dyskinetic mice. Biol Psychiatry. 2014; 75: 711-722.
- Kordower JH, Goetz CG, Chu Y, Halliday GM, Nicholson DA, Musial TF, et al. Robust graft survival and normalized dopaminergic innervation do not obligate recovery in a Parkinson disease patient. Ann Neurol 2017;81:46-57.