
Review Article
Austin J Nucl Med Radiother. 2014;1(1): 5.
Ionizing Radiation Induced Nitric Oxide Signaling
Rabender CS1*, Alam A1,2 and Mikkelsen RB1
1Department of Radiation Oncology, Virginia Commonwealth University, USA
2Department of Biochemistry, Virginia Commonwealth University, USA
*Corresponding author: Rabender CS, Department of Radiation Oncology, Virginia Commonwealth University, PO Box 980058 Richmond, VA 23298, USA
Received: August 25, 2014; Accepted: September 25, 2014; Published: September 27, 2014
Abstract
Radiotherapy is one of the most widely used forms of cancer therapy today used in the treatment of >60% of adult cancers. Treatment in many cases is limited as with other cancer therapeutics, due to normal tissue toxicity. Thus investigators are looking for ways to enhance the efficacy of radiation or to mitigate the damage to normal healthy tissues. Ionizing radiation stimulation of nitric oxide synthase activity has been studied extensively with conflicting results showing both cytotoxicity and cytoprotection. In this review experimental evidence is summarized suggesting that manipulation of nitric oxide signaling in combination with ionizing radiation, due to the dual nature of the cellular response to nitric oxide, has potential to enhance the anti-tumor efficacy of radiotherapy and mitigate damage to normal healthy tissue.
Keywords: Ionizing radiation; Nitric oxide synthase; ROS/RNS; Uncoupling
Abbreviations
BH2: Dihydrobiopterin; BH4: Tetrahydrobiopterin; cGMP: Cyclic Guanosine Monophosphate; DSB: Double Strand Breaks; EGFR: Epidermal Growth Factor Receptor; eNOS: Enothelial Nitric Oxide Synthase; GTPCH-I: Guanidine Triphosphate Cyclohydrolase I; HIF1-α: Hypoxia Inducible Factor 1-Alpha; iNOS: Inducible Nitric Oxide Synthase; IR: Ionizing Radiation; nNOS: Neuronal Nitric Oxide Synthase; NO: Nitric Oxide; O2: Superoxide; ONOO-: Peroxynitrite; PKG: Protein Kinase G; RNS: Reacitve Nitrogen Species; ROS: Reactive Oxygen Species; sGC: Soluble Guanylate Cyclase; VEGFR: Vascular Endothelial Growth Factor Receptor
Introduction
Ionizing Radiation (IR) is utilized in the treatment of a variety of tumors including breast, colon, lung and prostate. Exposure of mammalian cells to a clinically relevant dose of IR of approximately 2 Gy produces about 3000 DNA lesions: 850 pyrimidine lesions, 450 purine lesions, 1000 single strand breaks and 40 double strand breaks [1]. A hallmark of ionizing radiation is the formation of clustered damage sites, which include double strand breaks, characterized by two or more lesions within one or two helical turns of the DNA. These sites are thought to be the most cytotoxic lesions induced by IR [2,3]. DNA damage sensors within the nucleus detect this damage and initiate signal transduction pathways resulting in activation of cell cycle checkpoints and DNA damage repair. The cell's response involves a number of proteins including, but not limited to, ATM/ ATR, DNA-dependent protein kinase, Chk 1/2 and p53, as well as the generation of reactive oxygen species/reactive nitrogen species (ROS/ RNS), much of which may be attributable to the activation of nitric oxide synthase (NOS) [4-6].
Nitric Oxide (NO) is a highly diffusible regulator of several physiological processes; playing major roles in vasodilation, neurotransmission and the immune response. NO is produced in cells by Nitric Oxide Synthases (NOS). NOS are a group of calcium/ calmodulin responsive enzymes (eNOS (NOS III), nNOS (NOSI), and iNOS (NOSII)) that catalyze the production of NO (and L-citrulline) through the oxidation of L-arginine. NO has been shown in numerous investigations to be involved in the cellular response to IR. Leach et al. (2002) demonstrated low doses of IR activate a Ca2+ dependent NOS, while many others have shown up-regulation of iNOS in a wide range of tumor cells and tissues (glioblastoma, breast, head and neck) post IR exposure [7,8].
At low concentrations, << 1μM, NO binds to the heme-containing Soluble Guanylate Cyclase (sGC) resulting in the formation of cGMP leading to Protein Kinase G (PKG) activation. The binding of NO to the heme moiety of sGC is a direct effect of NO, but indirect effects of NOS activation are also observed. The indirect effects occur through the generation of ROS/RNS, often at much higher concentrations of NO, and occur through the interaction of NO with ROS, such as superoxide (O2-), generating different RNS. As NO is relatively stable and can diffuse readily throughout the cell, formation of RNS may be significant in the areas of greatest ROS generation; such as, the mitochondria and the plasma membrane near NADPH oxidases. Biologically relevant signaling from ROS/RNS has been demonstrated to occur through protein S-nitrosylation and tyrosine nitration [9- 11].
While numerous studies have demonstrated the antitumor effects of NO, it also promotes angiogenesis and metastasis suggesting that the concentration of NO, location of NO, whether it is an endogenous vs. exogenous source, and the tumor microenvironment, may determine the eventual cellular response. In this review, we will address the role NO plays in the efficacy of radiotherapy in terms of the mode of cell death, the evasion of treatment (radioresistance), involvement in tumor regeneration, and use as a radiosensitizer as well as a possible mechanism for the apparent conflicting results seen in previous studies.
Ionizing radiation induced NO promotes tumor cell toxicity
NO (or RNS) generated by ionizing radiation has been demonstrated to activate a number of stress proteins, including MAPK and JNK [12,13]. At low doses of IR or with low concentrations of RNS donors, activation of these pathways has been shown to be cytoprotective. We will discuss this more in the next section. At much higher levels of NO/RNS achieved with induction of iNOS expression or treatment with high levels of NO/RNS donors, cell damaging effects are seen [14,15]. It has been demonstrated that the cytotoxic effects of NO/RNS, at least in part, are due to their direct interaction with DNA and lipids producing DNA and lipid radicals leading to cell cycle arrest and apoptosis [16].
Numerous studies have shown that the biological effects of IR are not only a result of the irradiated cell, but also the neighboring un-irradiated cells. Results show that cells in the vicinity of the irradiated cells, bystander cells, respond in a similar fashion to the irradiated cell [17]. Not only are the irradiated tumor cells able to generate NO, but macrophages, being radioresistant, survive, get activated, and produce large amounts of NO [18,19].
Sokolov et al (2005) reported that irradiation of target cells induced γ-H2AX foci, a measure of double strand breaks (DSB) and DSB repair proteins, p53 ATM, Mre11, Rad50 and Nbs1, in bystander cell populations. The mechanism for the DSBs was not elucidated; however, pretreatment with the NO scavenger c-PTIO and Aminoguanidine (AG), a NOS inhibitor, abolished the effect [20]. Similar results have been published demonstrating the bystander effect in human glioblastoma T98G cells, where non-irradiated cells showed micronuclei induction by a process that was also blocked by c-PTIO and AG [21]. Shao et al (2003) went further to demonstrate that both NO and TGF-β1 are involved in the bystander effect in glioma cells. Treatment of the cells with AG reduced TGF-β1 to control levels, suggesting that these two factors are not independent. Further evidence for the involvement of TGF-β1 in the bystander effect has been shown by Arnold et al (1999). In this study they showed that the conditioned media from MCF-7 and MDA-MB-231 cells contained two-fold more TGF-β1 than that of un-irradiated cells [22].
Others have cited the necessity for NFκB induced iNOS resulting in elevated COX-2 expression as being involved in the radiation induced bystander effect [23]. The common denominator in all studies investigating the bystander effect is elevated NOS activity, but the mechanistic details are lacking. Future studies are needed to further elucidate the mechanism and the clinical relevance of the bystander effect.
Nitric oxide and nitric oxide synthase activity as a radiosensitizer
A variety of factors likely play a role in determining the therapeutic outcome of IR including hypoxia and the tumor vasculature. Studies have demonstrated significantly reduced radiosensitivity in tumor cells under conditions of low oxygen [24-27]. NO may increase tumor blood flow and tumor oxygenation, enhancing radiosensitivity of tumors [28,29] however the exact mechanism is unclear [24]. There is substantial evidence in multiple different tumor types showing increased radiosensitivity in tumors treated with NO donors [30-35]. Similar evidence suggests increased NOS activity may also increase radiosensitization as well [36-39].
Evidence to the contrary has been published as well. Our group and others have shown NOS inhibition can sensitize tumors to radiation [40-42]. Treatment of squamous cell carcinoma xenografts with the combination of L-NNA and radiation decreased tumor blood flow, leading to decreased growth in tumor cells, increased cytotoxicity of tumor cells as well as prolonged survival in mice [40]. NO and NOS activity may also play a dichotomous role in tumor cells and cells in the surrounding microenvironment. All of these studies illustrate the complexity in drawing conclusions in the actions of NO and NOS, either as a product of IR or in combination with IR.
Radiation-induced NOS activity is radioprotective in tumor cells
Although IR activates pathways leading to apoptosis and cell death, there is increasing evidence, at least in a subset of tumor cells, that radiation can also activate proliferative and pro-survival pathways. Our group and others have shown activation of the epidermal growth factor receptor (EGFR) signaling pathway after radiation, a protective mechanism, potentially leading to radio-resistance [43-47]. Lee et al (2008) showed this radiation induced activation of EGFR was dependent on NOS [48]. A potential mechanism for the activation of EGFR may be the cysteine oxidation of SHP2 protein phosphatase, shown to dephosphorylate Tyr992/1173 on EGFR. Studies have demonstrated that Cys453 on SHP2 is S-nitrosylated by a mechanism dependent on the production of reactive oxygen and nitrogen species via NOS, and inhibited post IR [10,49].
Hypoxia-inducible factor 1-alpha (HIF1α) can also be stabilized via NO/ROS-dependent cysteine oxidation leading to the activation of pro-survival and proliferative pathways [50,51]. The activation of HIF1α was also reported to cause the adaptation of glioma tumors to a more radio-resistant phenotype [52]. Matsumoto et al (2007) showed in human glioblastoma cells, treatment with exogenous NO as well as low dose IR contributed to a radioadaptive response. This radioadaptation was dependent on iNOS activity and levels [53].
Our lab has also demonstrated low dose IR and peroxynitrite activates NF-κB. Low dose IR-induced nitration of Tyr181 of IκBα causes IκBα to dissociate from NF-κB, activating the transcription factor. Inhibition of NOS activity with the non-specific NOS inhibitor, L-NNA, blocked nitration of IκBα and NF-κB activation [9]. This evidence suggests a combination of NO plus other ROS/RNS leads to an oxidative environment activating cytoprotective pathways decreasing the efficacy of radiation. A separate study showed that treatment of MCF-10A cells with low levels of RNS donors or co-culturing with activated macrophages results in the Tyr nitration and stimulation of PP2A activity leading to the down-regulation of BRCA1. A consequence is the reduction in homologous recombination DNA repair and enhanced non-homologous end-joining repair thereby promoting chromosomal instability, a hallmark of tumor progression [54]. The above evidence suggests that ROS/RNS may have multiple roles in tumor cell repopulation and acquired radioresistance but the actual mechanisms remain to be determined.
Radiation effects on surrounding cells in the tumor microenvironment
Radiation also activates pro- and anti-survival pathways in surrounding stromal cells such as fibroblasts and endothelial cells in the tumor microenvironment. These activated stromal cells provide cytokines and growth factors necessary for the tumor cells to survive radiation. Sonveaux et al (2003) demonstrated that radiation increased migration and capillary formation of endothelial cells in vitro as well as enhanced angiogenesis in matrigel plug assays (ex vivo). Treatment with the NOS inhibitor, L-NAME, abolished this radiation induced angiogenic effect [55]. Unpublished data in our lab demonstrates very similar results with 2H11 immortalized mouse tumor endothelial cells. In these experiments L-NNA blocked IR-induced migration of 2H11 cells, suggesting a possible mechanism for repopulation and angiogenesis post IR leading to radioresistance of tumor cells. IR has also been shown to activate vascular endothelial growth factor receptor (VEGFR) in multiple tumor types, enhancing post-irradiation angiogenesis [56,57]. A related phenomenon is that of irradiation induced angiosarcomas. Angiosarcomas are tumors derived from endothelial cells which have activated VEGFR and HIF1α signaling. These types of tumors typically arise after breast irradiation but can be found in many different sites as well as different tumor types such as meningiomas and sarcomas [58,59]. As mentioned above, VEGFR, EGFR and HIF1α all can be activated post irradiation via a mechanism dependent on ROS/RNS and NOS activity. Radiation also activates NF-κB and IL-6 production in mast cells and fibroblasts [60,61]. The activation of these cells enhances the inflammatory/pro-survival microenvironment of tumors, potentially leading to decreased efficacy of IR [62,63].
NOS uncoupling in tumor cells
Important questions that arise from the above discussion on the dual nature of NO mediated cellular responses to IR are what is the mechanism(s) that account for this duality in responses and can this mechanism(s) be therapeutically manipulated to enhance the efficacy of radiotherapy? An area of NO biosynthesis that has yet to be explored in great detail in tumor biology is NOS uncoupling. The synthesis of NO occurs through NOS dimers and requires the substrate arginine along with NADPH and molecular oxygen (O2) as co-substrates. Tetrahydrobiopterin (BH4), FAD and FMN are required cofactors [64]. Tetrahydrobiopterin is a necessary cofactor for the production of NO from NOS. Evidence has shown that NOS's can generate O2- under certain pathophysiological conditions and current research indicates that the level of BH4 is important in regulating the balance of O2- and NO produced by NOS. A BH4 molecule binds in the oxygenase domain of each NOS monomer resulting in two BH4 molecules in the active dimer. In conditions where BH4 levels are low, electron transfer in the active site of the enzyme becomes uncoupled from L-arginine oxidation resulting in the production of O2- instead of NO [65,66]. The uncoupled enzyme therefore becomes a "peroxynitrite synthase", which is produced rapidly by the reaction of O2- with NO produced in the same area.
In diabetes, hypertension and atherosclerosis loss of NO production is a common feature accounting in part for the endothelial dysfunction associated with these inflammatory diseases. Recent experiments showed that under certain inflammatory conditions the cofactor BH4 is limiting and that this results in reduced NO bioavailability. Low levels of BH4 can be a result of the low levels of GTP cyclohydrolase-I (GTPCH-I), the rate-limiting enzyme in the production of BH4, or through direct oxidation of BH4 to dihydrobiopterin (BH2) in the face of enhanced ROS [67,68]. Evidence has shown that NOS's have an equal affinity for BH4 and BH2 but when BH2 is bound the NOS dimer is unstable and O2- production dominates [69].
It has been demonstrated that ischemia reperfusion injury results in BH4 oxidation and it is ameliorated by exogenous BH4, suppressing NOS-derived superoxide [70]. A cells response to an IR event has been demonstrated to be similar in terms of the inflammatory response to that seen after vascular injury during ischemia reperfusion. Indeed, Berbee et al (2011) showed that mice exposed to 8.5Gy total body irradiation (TBI) displayed significantly decreased BH4 (pmol/ mg protein) at 3.5 and 7 days post IR [71]. BH4 is currently being evaluated for protection from post-irradiation vascular oxidative stress [72]. Our lab has evaluated the BH4 precursor, Sepiapterin (SP), in the DSS/AOM induced mouse model of colorectal cancer. Here we demonstrated that animals treated orally with SP showed decreased tumor formation when given SP in conjunction with DSS and AOM. A hallmark of the tumors generated by this model was a reduced BH4:BH2 ratio compared to normal colon tissue, which we were able to increase with SP in the drinking water [73]. We also observed increased levels of cGMP with SP treatment sensitive to inhibitors of the NO-dependent sGC. It has been shown that metastatic breast, colon and lung cancers have increased levels of PDE 2 and/or PDE 5 compared to normal tissue [74]. An alternative approach to stimulating cGMP production through exogenous activators of sGC is through inhibition of PDE's that break down cGMP. Exisulind , sulindac sulfone (a metabolite of the NSAID sulindac), has been shown to have pro-apoptotic effects in SW480 and HT29 cells by inhibiting PDE's leading to elevated cGMP [75,76].
Given that NO signaling in cells is mediated in large part by the generation of ROS/RNS, whether or not NOS is coupled can have significant effects on tumor cell signaling and response to IR. When NOS is fully coupled, NO dependent signaling pathways such as sGC and PKG dominate; whereas when NOS is uncoupled, ONOO- and other ROS/RNS signaling pathways dominate (Figure 1). As discussed earlier, depending on the cellular environment and concentration, the latter may actually be cytoprotective. The state of NOS coupling may be the reason for the paradoxical effects of NO seen in irradiated tumors.
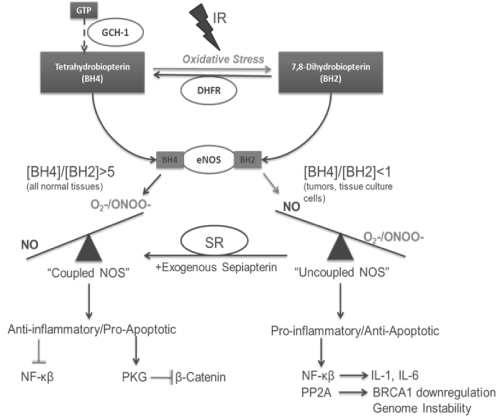
Figure 1 : Impact of NOS uncoupling on radiation induced NO Signaling.
Conclusion
In this short review, we have attempted to provide experimental evidence highlighting the role of NO in the cellular fate after exposure to an ionizing event as well as evidence suggesting the potential for manipulating NO signaling in combination with radiotherapy. NO can induce cellular damage by direct interaction with DNA, can protect from radiation-induced cell death and apoptosis by scavenging free radicals and inhibiting caspase activity as well as induction of a host of protective pathways through RNS signaling.
Further research is needed to determine the exact role of NO in the bystander effect as well as the therapeutic potential of NO as a radiosensitizer. Of particular interest to our lab is that radiation- induced ROS may decrease the cellular BH4 levels, leading to a prolonged stress response or even further activation of cytoprotective mechanisms. Future studies are needed to determine the exact role radiation plays in BH4 levels and NOS uncoupling in tumor cells during radiotherapy with the idea of continuing to characterize the dual nature of NO and the conflicting results observed.
References
- Ward JF. DNA damage as the cause of ionizing radiation-induced gene activation. Radiat Res. 1994; 138: S85-88.
- Sutherland BM, Bennett PV, Sidorkina O, Laval J. Clustered damages and total lesions induced in DNA by ionizing radiation: oxidized bases and strand breaks. Biochemistry. 2000; 39: 8026-8031.
- Sutherland BM, Bennett PV, Sidorkina O, Laval J. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc Natl Acad Sci U S A. 2000; 97: 103-108.
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996; 274: 1664-1672.
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000; 287: 1824-1827.
- Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998; 282: 1893-1897.
- Leach JK, Black SM, Schmidt-Ullrich RK, Mikkelsen RB. Activation of constitutive nitric-oxide synthase activity is an early signaling event induced by ionizing radiation. J Biol Chem. 2002; 277: 15400-15406.
- Kim RK, Suh Y, Cui YH, Hwang E, Lim EJ, Yoo KC, et al. Fractionated radiation-induced nitric oxide promotes expansion of glioma stem-like cells. Cancer Sci. 2013; 104: 1172-1177.
- Yakovlev VA, Barani IJ, Rabender CS, Black SM, Leach JK, Graves PR, et al. Tyrosine nitration of IkappaBalpha: a novel mechanism for NF-kappaB activation. Biochemistry. 2007; 46: 11671-11683.
- Barrett DM, Black SM, Todor H, Schmidt-Ullrich RK, Dawson KS, Mikkelsen RB. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem. 2005; 280: 14453-14461.
- Marshall HE, Hess DT, Stamler JS. S-nitrosylation: physiological regulation of NF-kappaB. Proc Natl Acad Sci U S A. 2004; 101: 8841-8842.
- Kavanagh BD, Dent P, Schmidt-Ullrich RK, Chen P, Mikkelsen RB. Calcium-dependent stimulation of mitogen-activated protein kinase activity in A431 cells by low doses of ionizing radiation. Radiat Res. 1998; 149: 579-587.
- Schmidt-Ullrich RK, Dent P, Grant S, Mikkelsen RB, Valerie K. Signal transduction and cellular radiation responses. Radiat Res. 2000; 153: 245-257.
- Bian K, Ghassemi F, Sotolongo A, Siu A, Shauger L, Kots A, et al. NOS-2 signaling and cancer therapy. IUBMB Life. 2012; 64: 676-683.
- Kielbik M, Klink M, Brzezinska M, Szulc I, Sulowska Z. Nitric oxide donors: spermine/NO and diethylenetriamine/NO induce ovarian cancer cell death and affect STAT3 and AKT signaling proteins. Nitric Oxide. 2013; 35: 93-109.
- Tamir S, Burney S, Tannenbaum SR. DNA damage by nitric oxide. Chem Res Toxicol. 1996; 9: 821-827.
- Mothersill C, Seymour C. Medium from irradiated human epithelial cells but not human fibroblasts reduces the clonogenic survival of unirradiated cells. Int J Radiat Biol. 1997; 71: 421-427.
- Ghosh S, Maurya DK, Krishna M. Role of iNOS in bystander signaling between macrophages and lymphoma cells. Int J Radiat Oncol Biol Phys. 2008; 72: 1567-1574.
- Jiang H, De Ridder M, Verovski VN, Sonveaux P, Jordan BF, Law K, et al. Activated macrophages as a novel determinant of tumor cell radioresponse: the role of nitric oxide-mediated inhibition of cellular respiration and oxygen sparing. Int J Radiat Oncol Biol Phys. 2010; 76: 1520-1527.
- Sokolov MV, Smilenov LB, Hall EJ, Panyutin IG, Bonner WM, Sedelnikova OA. Ionizing radiation induces DNA double-strand breaks in bystander primary human fibroblasts. Oncogene. 2005; 24: 7257-7265.
- Shao C, Stewart V, Folkard M, Michael BD, Prise KM. Nitric oxide-mediated signaling in the bystander response of individually targeted glioma cells. Cancer Res. 2003; 63: 8437-8442.
- Arnold SF, Tims E, Bluman EM, McGrath BE. Regulation of transforming growth factor beta1 by radiation in cells of two human breast cancer cell lines. Radiat Res. 1999; 152: 487-492.
- Zhou H, Ivanov VN, Gillespie J, Geard CR, Amundson SA, Brenner DJ, et al. Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc Natl Acad Sci U S A. 2005; 102: 14641-14646.
- Oronsky BT, Knox SJ, Scicinski JJ. Is Nitric Oxide (NO) the Last Word in Radiosensitization? A Review. Transl Oncol. 2012; 5: 66-71.
- Stevens G, Joiner M, Joiner B, Johns H, Denekamp J. Radiosensitization of mouse skin by oxygen and depletion of glutathione. Int J Radiat Oncol Biol Phys. 1995; 33: 399-408.
- Watts ME, Hodgkiss RJ, Jones NR, Fowler JF. Radiosensitization of Chinese hamster cells by oxygen and misonidazole at low X-ray doses. Int J Radiat Biol Relat Stud Phys Chem Med. 1986; 50: 1009-1021.
- Alper T. Adding two components of radiosensitization by oxygen. Int J Radiat Biol Relat Stud Phys Chem Med. 1984; 46: 569-585.
- Jordan BF, Misson P, Demeure R, Baudelet C, Beghein N, Gallez B. Changes in tumor oxygenation/perfusion induced by the no donor, isosorbide dinitrate, in comparison with carbogen: monitoring by EPR and MRI. Int J Radiat Oncol Biol Phys. 2000; 48: 565-570.
- Jordan BF, Sonveaux P, Feron O, Grégoire V, Beghein N, Gallez B. Nitric oxide-mediated increase in tumor blood flow and oxygenation of tumors implanted in muscles stimulated by electric pulses. Int J Radiat Oncol Biol Phys. 2003; 55: 1066-1073.
- De Ridder M, Verellen D, Verovski V, Storme G. Hypoxic tumor cell radiosensitization through nitric oxide. Nitric Oxide. 2008; 19: 164-169.
- Gao X, Saha D, Kapur P, Anthony T, Livingston EH, Huerta S. Radiosensitization of HT-29 cells and xenografts by the nitric oxide donor DETANONOate. J Surg Oncol. 2009; 100: 149-158.
- Griffin RJ, Makepeace CM, Hur WJ, Song CW. Radiosensitization of hypoxic tumor cells in vitro by nitric oxide. Int J Radiat Oncol Biol Phys. 1996; 36: 377-383.
- Janssens MY, Verovski VN, Van den Berge DL, Monsaert C, Storme GA. Radiosensitization of hypoxic tumour cells by S-nitroso-N-acetylpenicillamine implicates a bioreductive mechanism of nitric oxide generation. Br J Cancer. 1999; 79: 1085-10859.
- Policastro L, Duran H, Henry Y, Molinari B, Favaudon V. Selective radiosensitization by nitric oxide in tumor cell lines. Cancer Lett. 2007; 248: 123-130.
- Su X, Takahashi A, Kondo N, Nakagawa Y, Iwasaki T, Guo G, et al. Nitric oxide radical-induced radioadaptation and radiosensitization are G2/M phase-dependent. J Radiat Res. 2011; 52: 609-615.
- Berge DL, De Ridder M, Verovski VN, Janssens MY, Monsaert C, Storme GA. Chronic hypoxia modulates tumour cell radioresponse through cytokine-inducible nitric oxide synthase. Br J Cancer. 2001; 84: 1122-1125.
- De Ridder M, Van Esch G, Engels B, Verovski V, Storme G. Hypoxic tumor cell radiosensitization: role of the iNOS/NO pathway. Bull Cancer. 2008; 95: 282-291.
- McCarthy HO, Worthington J, Barrett E, Cosimo E, Boyd M, Mairs RJ, et al. p21((WAF1))-mediated transcriptional targeting of inducible nitric oxide synthase gene therapy sensitizes tumours to fractionated radiotherapy. Gene Ther. 2007; 14: 246-255.
- Worthington J, Robson T, O'Keeffe M, Hirst DG. Tumour cell radiosensitization using constitutive (CMV) and radiation inducible (WAF1) promoters to drive the iNOS gene: a novel suicide gene therapy. Gene Ther. 2002; 9: 263-269.
- Cardnell RJ, Mikkelsen RB. Nitric oxide synthase inhibition enhances the antitumor effect of radiation in the treatment of squamous carcinoma xenografts. PLoS One. 2011; 6: e20147.
- Liebmann J, DeLuca AM, Coffin D, Keefer LK, Venzon D, Wink DA, et al. In vivo radiation protection by nitric oxide modulation. Cancer Res. 1994; 54: 3365-3368.
- Babicová A, Havlínová Z, Hroch M, Rezácová M, Pejchal J, Vávrová J, et al. In vivo study of radioprotective effect of NO-synthase inhibitors and acetyl-L-carnitine. Physiol Res. 2013; 62: 701-710.
- Contessa JN, Abell A, Mikkelsen RB, Valerie K, Schmidt-Ullrich RK. Compensatory ErbB3/c-Src signaling enhances carcinoma cell survival to ionizing radiation. Breast Cancer Res Treat. 2006; 95: 17-27.
- Contessa JN, Hampton J, Lammering G, Mikkelsen RB, Dent P, Valerie K, et al. Ionizing radiation activates Erb-B receptor dependent Akt and p70 S6 kinase signaling in carcinoma cells. Oncogene. 2002; 21: 4032-4041.
- Lammering G, Hewit TH, Hawkins WT, Contessa JN, Reardon DB, Lin PS, et al. Epidermal growth factor receptor as a genetic therapy target for carcinoma cell radiosensitization. J Natl Cancer Inst. 2001; 93: 921-929.
- Lammering G, Hewit TH, Valerie K, Contessa JN, Amorino GP, Dent P, et al. EGFRvIII-mediated radioresistance through a strong cytoprotective response. Oncogene. 2003; 22: 5545-5553.
- Schmidt-Ullrich RK, Contessa JN, Lammering G, Amorino G, Lin PS. ERBB receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003; 22: 5855-5865.
- Lee HC, An S, Lee H, Woo SH, Jin HO, Seo SK, et al. Activation of epidermal growth factor receptor and its downstream signaling pathway by nitric oxide in response to ionizing radiation. Mol Cancer Res. 2008; 6: 996-1002.
- Sturla LM, Alexander MS, Mikkelsen RB. Mechanism for radiation-induced activation of EGFR: S-nitrosylation and inhibtion of SHP-2. Cancer Research, 2007.
- Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, et al. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. 2007; 26: 63-74.
- Köhl R, Zhou J, Brüne B. Reactive oxygen species attenuate nitric-oxide-mediated hypoxia-inducible factor-1alpha stabilization. Free Radic Biol Med. 2006; 40: 1430-1442.
- Kim YH, Yoo KC, Cui YH, Uddin N, Lim EJ, Kim MJ, et al. Radiation promotes malignant progression of glioma cells through HIF-1alpha stabilization. Cancer Lett. 2014.
- Matsumoto H, Takahashi A, Ohnishi T. Nitric oxide radicals choreograph a radioadaptive response. Cancer Res. 2007; 67: 8574-8579.
- Yakovlev VA. Nitric oxide-dependent downregulation of BRCA1 expression promotes genetic instability. Cancer Res. 2013; 73: 706-715.
- Sonveaux P, Brouet A, Havaux X, Grégoire V, Dessy C, Balligand JL, et al. Irradiation-induced angiogenesis through the up-regulation of the nitric oxide pathway: implications for tumor radiotherapy. Cancer Res. 2003; 63: 1012-1019.
- Ai P, Ren ZG, Wang X, Wang SC, Li P. [Effect of irradiation on matrix metalloproteinases, vascular endothelial growth factor and microvessel density of mice bearing Lewis lung cancer]. Sichuan Da Xue Xue Bao Yi Xue Ban. 2009; 40: 632-635.
- Chung YL, Jian JJ, Cheng SH, Tsai SY, Chuang VP, Soong T, et al. Sublethal irradiation induces vascular endothelial growth factor and promotes growth of hepatoma cells: implications for radiotherapy of hepatocellular carcinoma. Clin Cancer Res. 2006; 12: 2706-2715.
- Anzalone CL, Cohen PR, Diwan AH, Prieto VG. Radiation-induced angiosarcoma. Dermatol Online J. 2013; 19: 2.
- Azzariti A, Porcelli L, Mangia A, Saponaro C, Quatrale AE, Popescu OS, et al. Irradiation-induced angiosarcoma and anti-angiogenic therapy: a therapeutic hope? Exp Cell Res. 2014; 321: 240-247.
- Blirando K, Hneino M, Martelly I, Benderitter M, Milliat F, François A. Mast cells and ionizing radiation induce a synergistic expression of inflammatory genes in endothelial cells by a mechanism involving p38alpha MAP kinase and (p65) NF-kappaB activation. Radiation research. 2012; 178: 556-567.
- Brach MA, Gruss HJ, Kaisho T, Asano Y, Hirano T, Herrmann F. Ionizing radiation induces expression of interleukin 6 by human fibroblasts involving activation of nuclear factor-kappa B. The Journal of biological chemistry. 1993; 268: 8466-8472.
- Kamochi N, Nakashima M, Aoki S, Uchihashi K, Sugihara H, Toda S, et al. Irradiated fibroblast-induced bystander effects on invasive growth of squamous cell carcinoma under cancer-stromal cell interaction. Cancer Sci. 2008; 99: 2417-2427.
- Hellevik T, Martinez-Zubiaurre I. Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front Oncol. 2014; 4: 1.
- Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994; 298 : 249-258.
- Vásquez-Vivar J, Kalyanaraman B, Martásek P. The role of tetrahydrobiopterin in superoxide generation from eNOS: enzymology and physiological implications. Free Radic Res. 2003; 37: 121-127.
- Vásquez-Vivar J, Kalyanaraman B, Martásek P, Hogg N, Masters BS, Karoui H, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998; 95: 9220-9225.
- Mitchell BM, Dorrance AM, Webb RC. GTP cyclohydrolase 1 downregulation contributes to glucocorticoid hypertension in rats. Hypertension. 2003; 41: 669-674.
- Zheng JS, Yang XQ, Lookingland KJ, Fink GD, Hesslinger C, Kapatos G, et al. Gene transfer of human guanosine 5'-triphosphate cyclohydrolase I restores vascular tetrahydrobiopterin level and endothelial function in low renin hypertension. Circulation. 2003; 108: 1238-1245.
- Crabtree MJ, Smith CL, Lam G, Goligorsky MS, Gross SS. Ratio of 5,6,7,8-tetrahydrobiopterin to 7,8-dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS. Am J Physiol Heart Circ Physiol. 2008; 294: H1530-1540.
- Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, et al. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci U S A. 2007; 104: 15081-15086.
- Berbee M, Fu Q, Boerma M, Pathak R, Zhou D, Kumar KS, et al. Reduction of radiation-induced vascular nitrosative stress by the vitamin E analog γ-tocotrienol: evidence of a role for tetrahydrobiopterin. Int J Radiat Oncol Biol Phys. 2011; 79: 884-891.
- Berbée M, Fu Q, Kumar KS, Hauer-Jensen M. Novel strategies to ameliorate radiation injury: a possible role for tetrahydrobiopterin. Curr Drug Targets. 2010; 11: 1366-1374.
- Cardnell RJ, Rabender CS, Ross GR, Guo C, Howlett EL, Alam A, et al. Sepiapterin ameliorates chemically induced murine colitis and azoxymethane-induced colon cancer. J Pharmacol Exp Ther. 2013; 347: 117-125.
- Singer AL, Sherwin RP, Dunn AS, Appleman MM. Cyclic nucleotide phosphodiesterases in neoplastic and nonneoplastic human mammary tissues. Cancer Res. 1976; 36: 60-66.
- Zhu B, Vemavarapu L, Thompson WJ, Strada SJ. Suppression of cyclic GMP-specific phosphodiesterase 5 promotes apoptosis and inhibits growth in HT29 cells. J Cell Biochem. 2005; 94: 336-350.
- Goluboff ET. Exisulind, a selective apoptotic antineoplastic drug. Expert Opin Investig Drugs. 2001; 10: 1875-1882.