
Case Report
Ann Nurs Res Pract. 2022; 7(1): 1050.
Pediatric Acute Megakaryoblastic Leukemia with T(4;11) (Q21;Q23)/MLL-AF4 Fusion Transcript: A Case Report
Li Zhang1#, Hanyi Hou2#, Xiurong Gao1, Muxia Yan1, Yanmei Li1 and Yiqian Wang2*
1Department of Hematology, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, China
2Department of Biochemistry and Molecular Biology, GMU-GIBH Joint School of Life Sciences, Guangzhou Medical University, Guangzhou, China
#These authors have contributed to this work
*Corresponding author: Yiqian Wang, Department of Biochemistry and Molecular Biology, GMU-GIBH Joint School of Life Sciences, Guangzhou Medical University, Guangzhou, 511436, China
Received: July 26, 2022; Accepted: August 17, 2022; Published: August 24, 2022
Abstract
Acute Megakaryoblastic Leukemia (AMKL) is a rare and biologically heterogenous subtype of Acute Myeloid Leukemia (AML) with a diverse cytogenetic profile. Mix-lineage leukemia (MLL) rearrangements are commonly found in infant Acute Lymphoblastic Leukemia (ALL) and childhood AML. However, the occurrence of MLL rearrangements in AMKL is very rare. Here, we report a pediatric case of AMKL with MLL- AF4 fusion derived from t(4;11) (q21;q23). To our knowledge, the case has not been reported from China. Our observation supports understanding of molecular diversity in AMKL. The varying molecular features of AMKL are warranted to fully understand to develop novel therapeutic strategies.
Keywords: Acute megakaryoblastic leukemia; MLL-AF4 fusion gene; t(4;11)(q21;q23)
Introduction
Acute Leukemia (AL) is characterized by the clonal expansion of Hematopoietic Stem Cells (HSCs) or progenitors, blocking their differentiation into multiple blood lineages [1,2]. AL is divided into Acute Lymphoblastic Leukemia (ALL) and Acute Myeloid Leukemia (AML), mainly based on the type of leukemic-initiating cell lineage [3]. In the past few decades, the French-American-British (FAB) Cooperative Group has classified AML on the basis of morphologic features and number of blast cells. Acute Megakaryoblastic Leukemia (AMKL), resulting from the malignant accumulation of progenitors in the megakaryocyte lineage, occurs predominantly in children and is recognized as AML-M7 according to FAB system [4,5]. AMKL is divided into two major subgroups: patients with Down syndrome (DS-AMKL) and patients without Down syndrome (non-DS-AMKL) with the outcome of the latter one is generally poor with worse prognosis [6,7].
Chromosomal rearrangement of 11q23 involving the mixedlineage leukemia (MLL) gene is commonly found in childhood ALL and AML [8]. The most common rearrangement disrupting MLL gene involves a reciprocal translocation between MLL and a partner gene, resulting in a chimeric protein composed of the N-terminus domain of MLL and C-terminus domain of the partner gene. AF4 at 4q21 is a common partner for MLL, and MLL- AF4 fusion gene is observed in about 10% of patients diagnosed with B-cell ALL [9]. Involvement of the MLL-AF4 fusion transcript in AML is rarely found. Here, we present the first case of a Chinese pediatric AMKL patient with MLLAF4 fusion gene at our institution.
Case Report
A 5-year-old boy presented with fever and hemorrhage was admitted to our hospital. Physical examination detected anemia and petechiae on the neck and chest, as well as hepatosplenomegaly. In addition, cervical, axillary, and inguinal lymph nodes were negligible. A complete blood count and peripheral smear showed a White Blood Cell (WBC) count of 6.7×109/L, with 9% mature granulocytes, 82% lymphocytes, 1% monocytes and 8% blasts. The hemoglobin concentration was 85g/L, and the platelet count was 45×109/L. The bone marrow aspiration showed myeloid hyperplasia with 2% myeloid cells, 1% erythroid cells, 6% lymphoid cells and 91% blasts. Moreover, the blast cells had pale blue-gray cytoplasm, round nuclei with reticulated chromatin, high Nucleus-to-Cytoplasm (N/C) ratio and pseudopod formation (Figure 1A). Auer rods were not present, and some blasts showed cytoplasmic blebs. Platelet shedding was observed. Cytochemical staining showed that the blasts negative peroxidase (POX) -staining, positive Periodic Acid-Schiff (PAS) staining, and demonstrated alpha-naphthyl acetate esterase (alpha- NAE) reactivity (Figure 1B-C). Flow cytometry showed that the blast cells were positive for CD7, CD9, CD13, CD33, CD34, CD36, CD41a, CD42b and CD61 (Figure 2). Based on the above clinical observations, a diagnosis of AMKL (AML-M7) was made. Furthermore, Chromosomal analysis of the Bone Marrow (BM) leukemic blasts was performed at initial diagnosis using standard G-banding methods. The cytogenetic result revealed an abnormal karyotype with 46, XY, t(4;11) (q21;q23), del(12)(p11)[9]/46, XY[1], which indicated that the patient did not have DS although with a typical t(4;11) translocation. Fluorescence in situ hybridization (FISH) analysis using the MLL dual color ‘break- apart’ probe confirmed the rearrangement of MLL gene (Figure 3A). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed to identify the partner gene fused to MLL. PCR primers and PCR protocols were adopted from Pallisgaard et al [10]. Appropriate positive and negative controls were also included. The PCR analysis confirmed the existence of the MLL-AF4 transcript (Figure 3B). The patient received chemotherapy according to Chinese Childhood AML Cooperative Study Group Protocol including daunomycin and cytarabine, and achieved a Complete Remission (CR). Thereafter, the patient received one course of consolidation chemotherapy (daunomycin, cytarabine and etoposide) and one course of intensification chemotherapy with high-dose cytarabine. Later, the patient was discharged from the hospital for personal reason.
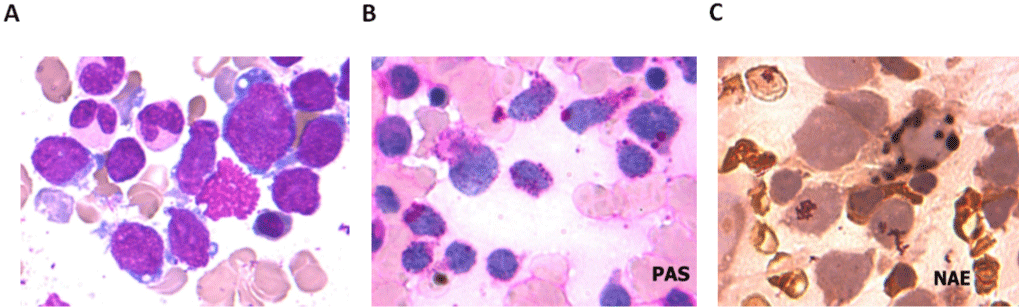
Figure 1: Morphological and cytochemical analyzes of leukemic cells from the patient. (A) May-Giemsa staining of BM cells at initial diagnosis. The blasts
showed cytoplasmic blebs and pseudopod formation (×1000). (B) Periodic acid-Schiff (PAS) staining showed block positive staining in the cytoplasm (×1000). (C)
The blasts revealed a granular pattern of alpha-NAE staining (×1000).
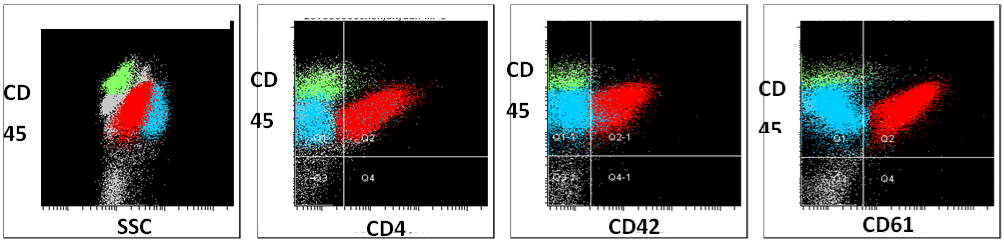
Figure 2: Representative flow cytometry data. FACS plots showing that the blast cells at diagnosis were positive for megakaryocytic markers including CD45,
CD41, CD42, and CD61.
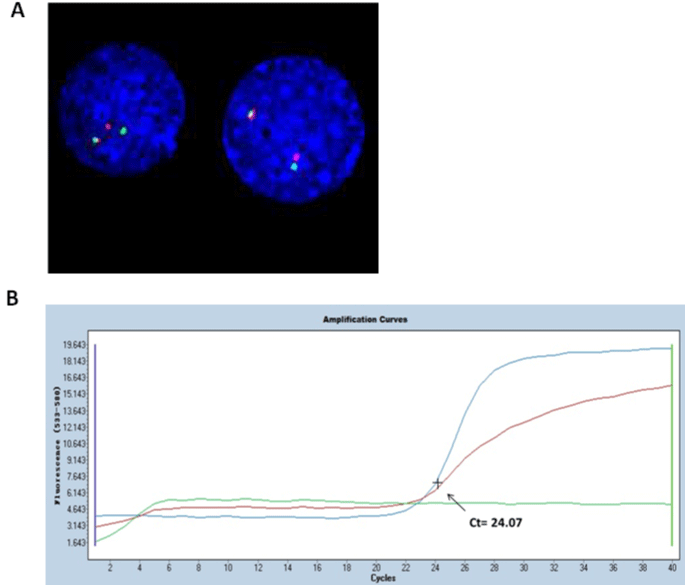
Figure 3: Cytogenetic analysis of leukemic blast cells at diagnosis. (A) MLL gene rearrangement detected by FISH. The positive signal mode of MLL generated
one red (1R), one green (1G) and one yellow fusion signal (1F). (B) qRT-PCR analysis result confirming MLL-AF4 fusion transcript identified in the blast. Green
curve: negative control; blue curve: patient; red curve: positive control. Curve time (Ct) value for positive reaction of MLL-AF4 fusion gene is equal to or less than 33.
Discussion
AMKL is caused by the malignant accumulation of progenitors in the megakaryocyte lineage [4]. Diagnosis of AMKL is established when more than 30% blasts in the Bone Marrow (BM) are leukemic cells according to FAB criteria [11]. Those blasts are identified as being of megakaryocyte lineage by use of monoclonal antibodies and electron microscopic platelet peroxidase reaction [11]. The diagnosis of AMKL may be difficult or confusing due to the difficulty in obtaining BM aspirates from the patient. Additionally, the morphology of leukemic blasts in AMLK can be similar to that of ALL, when some blasts show high chromatin density and scanty cytoplasm. However, megakaryoblasts in AMKL have some unique morphologic features, including cytoplasmic blebs, pseudopod formation and cytoplasmic projections resembling budding atypical platelets [12]. Moreover, cytochemical stains including PAS and NAE may also assist the diagnosis of AMKL. Ultimately, a variety of monoclonal antibodies can be used to identify megakaryoblasts, including CD41 (glycoprotein b/a), CD61 (glycoprotein a), and CD42b (glycoprotein b) [13].
Rearrangement of MLL gene, often seen in infant ALL and pediatric AML, is associated with a dismal prognosis, therapy refractoriness, Central Nervous System (CNS) infiltration, short latency, a high WBC count and a poor response to prednisone [14]. Chromosomal rearrangements of human MLL gene have been found to involved in reciprocal translocations, gene internal duplications, deletions or inversions on 11q, MLL gene insertions into other chromosomes, or the insertion of chromatin material into the MLL gene [15]. MLL rearrangement is very rarely seen in AMKL patients, and the most common recurrent cytogenetic abnormalities associated with MLL gene in AMKL were t(1;22)(p13;q13) and those involving 11q23, mostly t(9; 11)(p22;q23). The MLL-AF4 fusion in pediatric AMKL has been rarely reported [16]. Here, we describe the first reported case of pediatric AMKL with MLL-AF4 fusion in China.
Notably, the Event-Free Survival (EFS) for AMKL patients with MLL translocation is only 28.5%, and Overall Survival (OS) 32.4% [17]. A retrospective study by the European Group for Bone Marrow Transplantation reported a 3-year leukemia-free survival of 82 % after allogeneic hematopoietic stem cell transplant (allo-HSCT) in children with AMKL [18]. Furthermore, Garderet et al. reported that allo-HSCT treatment is superior to chemotherapy for improving outcomes for AML patients with MLL rearrangement [19]. Thus, allo-HSCT could be an alternative therapy for the patient in our case.
AMKL is characterized by leukemic megakaryoblasts that fail to undergo differentiation or terminal polyploidization [6]. Therefore, it is hypothesized that inducer of polyploidization and differentiation may exert anti-leukemic activity. Wen et al. showed that Aurora Kinase A (AURKA) negatively regulates polyploidization in AMKL, which is regarded as a potential therapeutic target [20]. Moreover, a more recent report indicated that targeting MLL-AF4 fusion protein also represents a promising therapeutic approach although the role of MLL fusions in AMKL is not well understood [21].
To our knowledge, this is the first report describing the molecular characteristics in MLL-AF4-associated AMKL in China. Our study highlights the cytogenetic heterogeneity of AMKL, and more importantly, provides new insights into the biology of AMKL, especially in pediatric patients.
Acknowledgements
This work was supported by the Guangzhou Municipal Science and Technology Project (202102021021), College Student Science and Technology Innovation Project of Guangzhou Medical University (2021A029), the Department of Education of Guangdong Province (2019KQNCX114), and the Guangzhou Municipal Science and Technology Project (202102010513).
Conflict of Interest
The authors declare no potential conflict of interest.
Author Contribution
Li Zhang and Yiqian Wang designed the study; Li Zhang, Hanyi Hou, Xiurong Gao, Muxia Yan, and Yanmei Li performed experiments. Li Zhang and Yiqian Wang wrote the manuscript. All authors reviewed the manuscript in its final form.
Ethics Statement
This study was approved by the Guangzhou Women and Children’s Medical Center Ethics Committee (ethics number: 2016103101).
Disclosure of Grants
This work was supported by the Guangzhou Municipal Science and Technology Project (202102021021), College Student Science and Technology Innovation Project of Guangzhou Medical University (2021A029), the Department of Education of Guangdong Province (2019KQNCX114), and the Guangzhou Municipal Science and Technology Project (202102010513).
References
- Brunning RD. Classification of acute leukemias. Seminars in diagnostic pathology. 2003; 20: 142-153.
- Tallman MS. Acute leukemias. Hematology/oncology clinics of North America. 2011; 25: xi-xii.
- Bennett JM, Catovsky D, Daniel M, Flandrin G, Galton DAG, Gralnick HR, et al. Proposals for the Classification of the Acute Leukaemias French- American-British (FAB) Co-operative Group. British Journal of Haematology. 1976; 33: 451-458.
- Lopez CK, Malinge S, Gaudry M, Bernard OA, Mercher T. Pediatric Acute Megakaryoblastic Leukemia: Multitasking Fusion Proteins and Oncogenic Cooperations. Trends in cancer. 2017; 3: 631-642.
- Catovsky D, Matutes E, Buccheri V, et al. A classification of acute leukaemia for the 1990s. Annals of hematology. 1991; 62: 16-21.
- Gruber TA, Downing JR. The biology of pediatric acute megakaryoblastic leukemia. Blood. 2015; 126: 943-949.
- Reinhardt D, Diekamp S, Langebrake C, Ritter J, Stary J, Dworzak M, et al. Acute megakaryoblastic leukemia in children and adolescents, excluding Down’s syndrome: improved outcome with intensified induction treatment. Leukemia. 2005; 19: 1495-1496.
- Winters AC, Bernt KM. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Frontiers in Pediatrics. 2017; 5.
- Godfrey L, Kerry J, Thorne R, Repapi E, Davies JOJ, Tapia M, et al. MLLAF4 binds directly to a BCL-2 specific enhancer and modulates H3K27 acetylation. Experimental Hematology. 2017; 47: 64-75.
- Pallisgaard N, Hokland P, Riishøj DC, Pedersen B, Jørgensen P. Multiplex reverse transcription-polymerase chain reaction for simultaneous screening of 29 translocations and chromosomal aberrations in acute leukemia. Blood. 1998; 92: 574-588.
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Criteria for the diagnosis of acute leukemia of megakaryocyte lineage (M7). A report of the French-American-British Cooperative Group. Annals of internal medicine. 1985; 103: 460.
- Rashidi A, Fisher SI. Acute megakaryoblastic leukemia. Blood. 2013; 122: 2537-2537.
- Wang L, Peters JM, Fuda F, Li L, Karandikar NJ, Koduru P, et al. Acute megakaryoblastic leukemia associated with trisomy 21 demonstrates a distinct immunophenotype. Cytometry Part B: Clinical Cytometry. 2015; 88: 244-252.
- Meyer C, Schneider B, Jakob S, Strehl S, Attarbaschi A, Schnittger S, et al. The MLL recombinome of acute leukemias. Leukemia. 2006; 20: 777-784.
- Meyer C, Hofmann J, Burmeister T, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013; 27: 2165-2176.
- Takita J, Motomura A, Koh K, Ida K, Taki T, Hayashi Y, et al. Acute megakaryoblastic leukemia in a child with the MLL-AF4 fusion gene. European Journal of Haematology. 2009; 83: 149-153.
- Inaba H, Zhou Y, Abla O, Adachi S, Auvrignon A, Beverloo HB, et al. Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood. 2015; 126: 1575-1584.
- Hahn AW, Li B, Prouet P, Giri S, Pathak R, Martin MG. Acute megakaryocytic leukemia: What have we learned. Blood reviews. 2016; 30: 49-53.
- Garderet L, Labopin M, Gorin N, Polge E, Baruchel A, Meloni G, et al. Hematopoietic stem cell transplantation for de novo acute megakaryocytic leukemia in first complete remission: a retrospective study of the European Group for Blood and Marrow Transplantation (EBMT). Blood. 2005; 105: 405- 409.
- Wen Q, Goldenson B, Silver SJ, Schenone M, Dancik V, Huang Z, et al. Identification of Regulators of Polyploidization Presents Therapeutic Targets for Treatment of AMKL. Cell. 2012; 150: 575-589.
- Marschalek R. MLL Leukemia and Future Treatment Strategies. Archiv der Pharmazie. 2015; 348: 221-228.