
Case Presentation
Austin J Obstet Gynecol. 2016; 3(3): 1065.
Hereditary Apert Syndrome: Case Report and Literature Review
Naveiro-Fuentes M¹*, Carrillo-Badillo MP¹, Culiañez-Casas M², Malde-CondE FJ¹ and Puertas A¹
¹Department of Gynecology and Obstetrics, Virgen de las Nieves Universitary Hospital, Spain
²Department of Radiology, Virgen de las Nieves Universitary Hospital, Spain
*Corresponding author: Naveiro-Fuentes M, Department of Gynecology and Obstetrics, Virgen de las Nieves Universitary Hospital, Granada, Spain
Received: October 07, 2016; Accepted: December 12, 2016; Published: December 14, 2016
Abstract
Apert syndrome is a rare genetic disorder caused by a mutation in the FGFR2 gene, and which is part of a suite of syndromes characterized by craniosynostosis or premature fusion of the cranial coronal sutures. Inheritance is autosomal dominant, although the syndrome generally occurs as a result of a de novo mutation. Patients with Apert syndrome have acrocephaly frequently associated with central nervous system disorders and symmetric syndactyly in their hands and feet. A suspected diagnosis is often based on second trimester ultrasonographic images, although the definitive diagnosis requires genetic testing to identify the mutation. We discuss a case of hereditary Apert syndrome, its etiology, clinical characteristics and modes of intrauterine diagnosis.
Keywords: Acrocephalosyndactilia; Craniosynostosis; Fibroblast growth factor type 2 receptor; Pregnancy; Prenatal ultrasonography
Abbreviations
CNS: Central Nervous System; FGFR2: Fibroblastic Growth Factor 2 Receptor; MRI: Magnetic Resonance Imaging studies
Introduction
Apert syndrome, also termed type 1 acrocephalosyndactyly, is a congenital disorder of genetic origin with an estimated prevalence of 6.5 to 15.5 cases per million live births [1]. Clinically, the syndrome is characterized by craniofacial abnormalities consisting mainly of coronal suture synostosis and maxillary hypoplasia, with symmetrical syndactyly in the hands and feet in which the three central digits are fused. Central Nervous System (CNS) anomalies may also be present, such as ventriculomegaly or agenesis of the corpus callosum, which lead to intellectual disability in approximately half of the patients. Other less frequent anomalies are cardiovascular or urogenital malformations, fusion of the cervical vertebrae and cleft palate [2,3].
The mode of inheritance is autosomal dominant, although the great majority of cases described to date are sporadic and have been associated with older age of the father [1,2]. The origin lies in a mutation of the genes that encode fibroblastic growth factor 2 receptor (FGFR2), localized on the long arm of chromosome 10 [4].
We describe a fetus diagnosed in week 32 of gestation as having hereditary Apert syndrome whose father also had this disorder. The mother provided her informed consent in writing for publication of information about this case.
Case Presentation
The mother was a 27-year-old white woman with three healthy children from a previous relationship. She had not received routine obstetric care for the pregnancy described here until week 30 of gestation, when ultrasonographic examination disclosed suspected lobar holoprosencephaly and polyhydramnios. She was then referred to the Fetal Medicine Unit of the Virgen de las Nieves University Hospital (Granada, Spain).
Her current partner and putative father of the fetus was a 38-yearold white man with no consanguineous relationship with the mother. Physical examination showed a high, prominent forehead, flat occiput and hypertelorism, and fusion of the three central fingers of both hands. On questioning about these malformations, he reported that they were due to an unidentified congenital infection. He had no other known malformations and no apparent intellectual disability.
In an obstetric ultrasonographic examination done in week 32, notable findings were moderate polyhydramnios with a pocket larger than 12cm and abnormal features in the cranial anatomy consisting of flat occiput, depressed nasal bridge, prominent forehead and closure of the coronal suture (Figure 1a). Fusion of the anterior horns and mild bilateral colpocephaly (dilatation of the trigones and occipital horns) were also seen (Figure 1b). The morphological appearance of the posterior fossa was normal.

Figure 1: (a): Ultrasonographic image at 32 weeks of gestation, showing a
flattened occiput, depressed nasal bridge and prominent forehead. (b):
Coronal view of the fetal skull showing fusion of the anterior horns in both
lateral ventricles.
Severe hand and foot malformations were evident, with severe syndactyly affecting all digits. The long bones were morphologically normal although small (below the 5th percentile).
Apert syndrome was suspected, and amniocentesis was done for karyotyping and examination for the presence of the FGFR2 mutation.
Magnetic Resonance Imaging studies (MRI) were requested for complete study of the cerebral malformations. The father was also advised that genetic studies would be informative in light of the suspicion that his malformations were due to Apert syndrome rather than congenital infection.
Fetal MRI confirmed the findings and further disclosed anatomical abnormalities in the head consistent with turribrachicephaly, which included enlarged biparietal diameter, shortened anteroposterior diameter and compensatory vertical development. Right occipital plagiocephaly due to local flattening of the cranial vault was also observed. These features were all indirect signs of craniosynostosis. Squaring of the anterior horns, possible fusion of the mammillary bodies, moderate ventriculomegaly (12mm) in the occipital horns, and a well developed posterior fossa with normal-appearing vermis and cerebellar hemispheres were also seen.
There was hypoplasia of some facial structures, with flattening of the forehead, a depressed nasal bridge, hypertelorism, shallow orbits and hypoplasia of the maxilla (Figure 2).
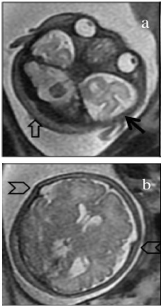
Figure 2: Magnetic resonance image (T2-weighted single-shot fast-spin
echo) of the fetus at 33 weeks of gestation in the axial plane.
(a) The transverse diameter of the skull is enlarged by the prominent temporal
bones (dark arrow), and the anteroposterior diameter is decreased due to
right occipital plagiocephaly (open arrow). Ocular hypertelorism is also
evident. (b) An image obtained at a higher plane shows fusion of the coronal
suture (arrowheads).
Genetic studies in both the father and the fetus disclosed a heterozygous c758C>G (p.Pro253Arg) mutation in the FGFR2 gene compatible with a diagnosis of Apert syndrome.
When informed of the results of these studies, the mother and her partner decided to have the pregnancy terminated legally at week 33 of gestation. Legal interruption of pregnancy is allowed in our country after the approval of the hospital ethics committee that evaluates the case and anomalies.
On macroscopic observation the fetus had characteristics compatible with Apert syndrome, including bilateral syndactyly in the hands and feet, midface hypoplasia and a prominent forehead (Figure 3). Autopsy confirmed the ultrasonographic findings.
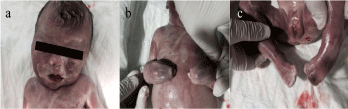
Figure 3: Gross examination on fetal autopsy confirmed midface hypoplasia and a prominent forehead (a) and syndactyly in the hands and feet (b,c).
Discussion
Apert syndrome belongs to a suite of syndromes characterized by craniosynostosis, and caused by mutations in genes of the FGFR2 family located on the long arm of chromosome 10 [2]. The incidence of Apert syndrome ranges in different studies from 6.5 to 15.5 cases per million live births, and represents 4.5% of all syndromes with craniosynostosis [1].
Most FGFR2 gene mutations are antisense mutations. Apert syndrome involves mainly two types of mutation that occur in 98% of all cases: p.Pro253Arg and p.Ser252Trp. The p.Pro253Arg mutation is reportedly associated with more pronounced syndactyly, whereas the p.Ser252Trp mutation leads to more severe facial malformations [5]. In the fetus we describe, the mutation involved was p.Pro253Arg, and the gross anatomical findings were consistent with the tendency for this form to be associated with more severe syndactyly given the complete fusion of all fingers in both hands (spade-type fusion) with no separation between the thumb or little finger (Figure 3b).
In most reports published to date, Apert syndrome was diagnosed in the third trimester [6,7], but thanks to advances in traditional and three-dimensional ultrasound methods, currently the diagnosis is often reached on the basis of ultrasound examination in the second trimester, at approximately week 20 [8-10]. In the case reported here, early diagnosis was not possible because the mother was not included in the public health prenatal care program during the early part of her pregnancy, a circumstance that ruled out the possibility of early detection of any malformations.
Prenatal ultrasound diagnosis is based on the detection of a characteristic triad: cranial abnormalities, midface hypoplasia and syndactyly in the hands and feet. Hypertelorism is also a characteristic of Apert syndrome and should alert obstetricians to the possibility of a syndrome involving craniosynostosis. Other associated anomalies are CNS, cardiac or urogenital malformations. In the case described here, the fetus had the characteristic malformations of Apert syndrome, and neither ultrasound nor autopsy detected malformations in other sites.
On ultrasound examination the fetus had a characteristic set of malformations (Figure 1), with a depressed nasal bridge, prominent forehead and protrusion of the lower jaw as a result of midface hypoplasia. These findings should alert clinicians to the possibility of Apert syndrome or some other syndrome involving craniosynostosis.
Magnetic resonance imaging can also play an important role in the diagnosis, mainly by documenting other CNS malformations. This technique can thus confirm or rule out the presence of certain abnormalities more reliably than ultrasound images can [8,9]. In the case reported here, MRI confirmed the presence of cranial malformations such as hypertelorism, brachicephaly, vertical orientation of the clivus, abnormal craniocervical junction angle and fusion of the mammillary bodies of the anterior horns. Thus MRI played a fundamental role as a complementary technique in addition to ultrasound examination in characterizing the cranial malformations (Figure 2) [8,11].
Three-dimensional ultrasound can be helpful in documenting syndactyly in the hands and feet [12,13]. In the fetus we examined, we visualized complete fusion of all fingers, which was subsequently confirmed on macroscopic examination (Figure 3).
When these malformations are seen, the clinician should suspect a craniosynostosis syndrome. Differential diagnosis should include Crouzon, Pfeiffer and Carpenter syndromes, which are also characterized by craniosynostosis and are also caused by a FGFR2 gene mutation. A definitive diagnosis may be impossible to reach on the basis of obstetric ultrasound examination alone. Crouzon syndrome is not characterized by hand or foot malformations, and facial malformations are less severe than in Apert syndrome. Carpenter syndrome is characterized by preaxial polydactyly, which is not a feature of Apert syndrome. Pfeiffer syndrome is also characterized by craniosynostosis, partial syndactyly and hypertelorism [2,4].
Given the similarities among these syndromes, the diagnosis must be confirmed with genetic testing to identify a mutation in the FGFR2 gene in the amniotic fluid or chorionic villi. If specific tests are not requested, the diagnosis is likely to be inaccurate since conventional karyotypes are normal and do not disclose anomalies that might raise concerns [8].
Although most cases described to date were caused by a de novo mutation [8,10], the case we report here is particularly interesting because the autosomal dominant mode of inheritance made hereditary Apert syndrome in this couple’s child a distinct possibility, yet the father was unaware of his own diagnosis.
People with the autosomal dominant type of inheritance have a 50% risk of passing on Apert syndrome to their children. Prospective parents can be offered several options including the use of donor semen, preimplantation genetic testing or natural conception with early diagnosis based on invasive testing during the first trimester.
Acknowledgment
We thank K. Shashok for translating the original manuscript into English.
References
- Tolarova MM, Harris JA, Ordway DE, Vargervik K. Birth prevalence, mutation rate, sex ratio, parents’ age, and ethnicity in Apert syndrome. Am J Med Genet. 1997; 72: 394-398.
- KL J. Craniosynostosis Syndromes. In: Jones KL JM, Del Campo M editor. Smith’s recognizable patterns of human malformation. Philadelphia: Elselvier. 2013; 536-537.
- OMIM. #101200 Apert Syndrome. 2014.
- GeneReview. FGFR-Related Craniosynostosis Syndromes. 2014.
- von Gernet S, Golla A, Ehrenfels Y, Schuffenhauer S, Fairley JD. Genotypephenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery. Clin Genet. 2000; 57: 137-139.
- Filkins K, Russo JF, Boehmer S, Camous M, Przylepa KA, Jiang W, et al. Prenatal ultrasonographic and molecular diagnosis of Apert syndrome. Prenat Diagn. 1997; 17: 1081-1084.
- Kaufmann K, Baldinger S, Pratt L. Ultrasound detection of Apert syndrome: a case report and literature review. Am J Perinatol. 1997; 14: 427-430.
- Giancotti A, D’Ambrosio V, De Filippis A, Aliberti C, Pasquali G, Bernardo S, et al. Comparison of ultrasound and magnetic resonance imaging in the prenatal diagnosis of Apert syndrome: report of a case. Childs Nerv Syst. 2014; 30: 1445-1448.
- David AL, Turnbull C, Scott R, Freeman J, Bilardo CM, van Maarle M, et al. Diagnosis of Apert syndrome in the second-trimester using 2D and 3D ultrasound. Prenat Diagn. 2007; 27: 629-632.
- Pi G, Zuniga A, Cervera J, Ortiz M. Prenatal diagnosis of Apert syndrome caused by de novo mutation in FGFR2 gene. An Pediatr (Barc). 2014; 80: 104-105.
- Weber B, Schwabegger AH, Vodopiutz J, Janecke AR, Forstner R, Steiner H. Prenatal diagnosis of apert syndrome with cloverleaf skull deformity using ultrasound, fetal magnetic resonance imaging and genetic analysis. Fetal Diagn Ther. 2010; 27: 51-56.
- Esser T, Rogalla P, Bamberg C, Kalache KD. Application of the threedimensional maximum mode in prenatal diagnosis of Apert syndrome. Am J Obstet Gynecol. 2005; 193: 1743-1745.
- Moes JA, Boyle JG, Flanagan JD, Mroch AR, Stein QP. Three-dimensional ultrasound to detect Apert syndrome and improve patient understanding. S D Med. 2011; 64: 125-127.