Case Report
Austin Oncol Case Rep. 2021; 4(1): 1014.
Complete Hematological Remission after Decitabine Treatment in a Patient with Congenital Agammaglobulinemia, FLT3- and TP53-Mutated Acute Myeloid Leukemia
Vigna E¹, Caracciolo D¹, Tenuta R², Martino E¹, Mendicino F¹, Caruso N¹, Greco F², Botta C¹, Morelli R³, Gentile M¹*
¹Hematology Unit AO of Cosenza, Cosenza, Italy
²Microbiology and Virology Unit, AO of Cosenza, Cosenza, Italy
³Internal Medicine Department, AO of Cosenza, Cosenza, Italy
*Corresponding author: Massimo Gentile, MD, Hematology Unit, AO of Cosenza, 87100 Cosenza, Italy
Received: April 12, 2021; Accepted: April 30, 2021; Published: May 06, 2021
Abstract
Older and unfit patients with Acute Myeloid Leukemia (AML), which are uneligible for standard induction therapy, have limited treatment options. The therapeutic approach in these cases is based on the use of hypomethylating agents, either decitabine or azacitidine, or Low-Dose Cytarabine (LDAC). However, despite the extensive use of these agents, there is no consensus regarding the extent of their efficacy, and clinical benefit deriving from their use is very modest. We present a case of FLT3- and TP53-mutated AML in an unfit patient with congenital agammaglobulinemia, responsive to single agent decitabine, with a response duration of over 20 months.
Keywords: AML; Decitabine; TP53-mutated; FLT-3 mutated
Introduction
Acute Myeloid Leukemia (AML) is a hematological malignancy characterized by abnormal proliferation of immature hematopoietic progenitor cells of the myeloid lineage. Importantly, no standard of care therapy is present for older or unfit patients and therefore outcomes are generally worse than in younger and fit patients [1].
In this context, therapy with hypomethylating agents (decitabine or azacytidine) as single agents or in combination with targeted therapies, has also been increasingly used in older or unfit AML patients. However, these therapeutic strategies in the older and unfit population, offer a modest survival advantage with a median OS ranging from 7.5 to 11 months [2]. Immunodeficiency increases the risk of malignancies, which occur earlier in life of these patients in respect to the general population. Congenital agammaglobulinemia is a Primary Immunodeficiency Disorder (PID) characterized by a severe reduction of all classes of serum immunoglobulins and by the absence of mature CD19+ B-cells. Although different malignancies have been observed in patients with congenital agammaglobulinemia [3], in this work we have presented the first reported case of FMSLike Tyrosine Kinase 3- (FLT3) and TP53-mutated AML in a young patient with congenital agammaglobulinemia. Furthermore, despite the poor prognosis of our clinical case, we here show the complete remission of AML after single agent decitabine treatment with duration of over 20 months.
Case Presentation
A 29-year-old woman, with a history of recurrent pulmonary infections and mental retardation due to agenesis of the corpus callosum, was followed for about 10 years by our Hematologic clinic for a congenital agammaglobulinemia, managed with intravenous immunoglobulin replacement therapy.
In June 2019 she went to the emergency room with fever without an apparent focus of infection. Laboratory tests revealed hyperleukocytosis [White Blood Cells count (WBC) of 64.2x103/ μl], anemia [hemoglobin (Hb) 8 gr/dL] and normal platelet count (PLT 153x103/μl). Morphologic analysis of peripheral blood smear revealed the presence of 70% myeloid blasts and the subsequent bone marrow (BM) aspiration confirmed the diagnosis of AML with 80% myeloid blasts. Flow-cytometry identified the presence of aberrant cells with the follow immunophenotype: MPO-, cyCD3-, cyCD79a-, TdT-, CD34+, CD117+/, CD33+, CD13+/-, HLADr+, CD38+, CD15+/-, CD11b-, CD16-, CD14-, CD64/+, CD4+/-, CD7+, CD56+. Cytogenetic analysis revealed a complex karyotype (45, XX,-5, der (6),-8,-12,-13,-14,+5der(?)[14]/46, XX[6]) (Figure 1) and bio-molecular analysis reported FLT3-ITD mutation with high allelic burden (>0,5) and TP53 mutation, while no mutations were found on NPM1 gene. Bone marrow aspiration confirmed also B-cell hypoplasia with significant reduction of CD19+ B-cells.
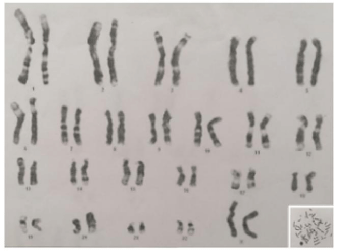
Figure 1: Cytogenetic analysis at the time of diagnosis.
The clinical presentation, the complex karyotype and the biomolecular features permitted us to classify the patient in a high-risk category, with a very poor prognosis. Initial cytoreduction was started with Hydroxyurea 3 g/day.
After 20 days from the start of cytoreduction, the hemogram showed a normalization of WBC (9.8x10³/μl) and the occurrence of thrombocytopenia (PLT 15x10³/μl). Hydroxyurea was stopped and, due to the unfit medical conditions of the patient, we decided to initiate treatment with decitabine (20 mg/m² for 5 consecutive days every 28 days).
After 21 days from the start of the first cycle of decitabine, the patient again showed hyperleukocytosis with WBC 51.2x10³/μl, anemia (Hb 8,4 gr/dl) and thrombocytopenia (PLT 21x10³/μl). At this point, hydroxyurea-based cytoreductive therapy was re-introduced. The patient was febrile again with the appearance of erythematous lesions on the left arm and daptomycin was started, with slight resolution of the thermal curve and of the cutaneous lesion.
After improvement of general clinical conditions, the second cycle of decitabine was started.
After 6 cycles of treatment with decitabine as single agent a significant hematological complete response was obtained without significant side effects. Indeed, in February 2020, the patient showed a normal hemogram with WBC 5.9x103/μl, Hb 12 gr/dl and PLT 158x103/μl. Consistently, BM aspiration reported <5% of blast cells, along with the restoration of normal hematopoiesis. Interestingly, while cytogenetic analysis reported the same complex karyotype of disease onset (45,XX,-5,der(6),-8,-12,-13,-14,+5der(?)[14]/46, XX[6]), bio-molecular analysis showed the presence of a FLT3-ITD mutation with low allelic burden (<0,5) (Figure 2) and reduction in variant allele frequency of TP53 mutation to levels of less than 5%. Most importantly the hematologic response was conserved for 20 months from the beginning of therapy.
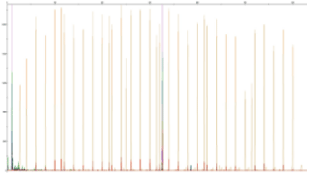
Figure 2: Pherogram showing low burden FLT3-ITD mutation (MUT/WT <0, 0409), as observed after decitabine treatment.
Discussion
Older AML patients or those with comorbidities and poor performance status at diagnosis are not eligible for intensive chemotherapy, due to increased treatment-related morbidity and mortality.
In this context, decitabine is a well-tolerated alternative to intensive chemotherapy, but offers a modest advantage.
On these premises, we presented an exceptional case of high risk FLT3- and TP53- mutated AML patient responder to single agent decitabine.
Different studies have suggested that certain subgroups of AML patients could benefit more from hypomethylating agents. For example, AML patients with complex karyotype, like those observed in our case report, had significant response rates to decitabine in phase II AML studies. Furthermore, it is well known that, although AML patients with TP53 mutations have very low response rates after standard cytotoxic therapy, all patients with a TP53 mutation are highly sensitive to decitabine treatment [4]; indeed, we observed a significant reduction of allele frequency of TP53 mutation after decitabine treatment.
Here we also reported a partial molecular response to decitabine, with FLT3 mutation allelic burden shifting from high to low after 6 cycles of therapy.
Interestingly, our findings could be consistent with recent experimental evidence which show that expression of FLT3 and its downstream targets were decreased in FLT3-ITD mutant cell lines treated with decitabine. Mechanistically decitabine-mediated upregulation of PU.1, led to downregulation of FLT3 which in turn triggers apoptosis of AML-mutated cells [5].
In conclusion, although further studies are needed to determine which subset of patients could benefit from single-agent decitabine therapy and, despite the short- lived remissions obtained by this therapeutic approach, our findings may provide a novel proof of concept for the treatment of FLT3-ITD and TP53 mutated AML unfit patients, highlighting at the same time, the feasibility of a safer therapeutic approach as bridge to allogeneic stem-cell transplantation.
Contributions
E.V., M.G. designed the study; E.M., E.V., C.B., N.C., F.G., R.T., R.M., D.C., F.M., M.G. analysed and interpreted data, and wrote the manuscript; all authors gave final approval for the manuscript.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
References
- Eleni LD, Nicholas ZC, Alexandros S. Challenges in treating older patients with acute myeloid leukemia. J Oncol. 2010; 2010: 943823.
- Stone A, Zukerman T, Flaishon L, Yakar RB, Rowe JM. Efficacy outcomes in the treatment of older or medically unfit patients with acute myeloid leukaemia: A systematic review and meta-analysis. Leuk Res. 2019; 82: 36-42.
- Satgé D. A Tumor Profile in Primary Immune Deficiencies Challenges the Cancer Immune Surveillance Concept. Front Immunol. 2018; 9: 1149.
- Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TTP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med. 2016; 375: 2023-2036.
- Hu X, Cai J, Zhu J, Lang W, Zhong J, Zhong H, et al. Modulation of FLT3 through decitabine-activated C/EBPa-PU.1 signal pathway in FLT3-ITD positive cells. Cell Signal. 2019; 64: 109409.