Case Report
Austin Oncol Case Rep. 2024; 7(1): 1022.
Two Brothers Diagnosed with Ewing Sarcoma: Unusual Case Report
Aytül T*; Gökalp RA; Betül BS
Department of Pediatric Oncology, Uludag University, Turkey
*Corresponding author: Aytül T Department of Pediatric Oncology, Uludag University, Gorukle/Nilüfer, Bursa/ Turkey. Tel: +905342321272 Email: aytultemuroglu@uludag.edu.tr
Received: April 17, 2024 Accepted: May 13, 2024 Published: May 20, 2024
Abstract
Ewing sarcoma is a childhood tumor involving bone and soft tissue. It is most commonly seen in the first two decades of life. Epidemiological studies continue to investigate the hereditary and environmental causes of this condition. It is considered non-familial. To date, six sibling cases have been reported in three publications, and all of these siblings were female. In this paper, we present the first cases of two male siblings with Ewing sarcoma in the literature.
Introduction
Ewing sarcoma is the second most common malignant tumor of bone after osteosarcoma in childhood. It is a very rare condition, being seen in three per million children [1]. It is slightly more common in boys than in girls [2]. It is most commonly located in the pelvis, femur, tibia, and humerus. Bone involvement is seen in 85% of cases and soft tissue in 15%. The mean age at the time of diagnosis is reported to be 15 years. Up to 25% of cases may present with metastasis at diagnosis [3]. The most common metastases are those of the lung, bone, and bone marrow. Survival has gradually increased over the years, with the 10-year survival rate reaching 63% in the presence of local disease [4].
Previous studies have reported that Ewing sarcoma is observed more frequently in families engaged in farming and in children with hernias [5,6]. With developments in molecular genetics, researchers have attempted to determine whether there is a genetic predisposition to this condition. In 85% cases of Ewing sarcoma, t(11;22)(q24;q12) somatic mutations have been reported [7,8]. Mutations in these genes result in the production of the EWS/ETS fusion protein, which functions as a transcription factor [9].
Studies have shown that P53 gene mutations are more common in Ewing sarcoma [10,11]. However, this condition has not been recognized as part of Li-Fraumeni syndrome [12]. Ewing sarcoma has been determined to be more common in some ethnicities and less common in Africans. It is considered to be non-familial [6].
In this paper, we present two brothers diagnosed with Ewing sarcoma, which, to the best of our knowledge, is the first report of its kind in the literature.
Case 1
A seven-year-old male patient presented to our hospital with complaints of weakness in the legs and an inability to walk for 20 days. He had no remarkable medical history. There was no consanguinity between his parents. His parents were both 34 years old. When his family history was examined, his grandmother had been diagnosed with breast cancer at the age of 55, and his grandfather had been diagnosed with lung cancer at the age of 60. The family was not engaged in farming. The mother did not smoke or consume alcohol. The father smoked at home. An ablation procedure had been performed on the mother when she was 30 years due to arrhythmia. In the physical examination of the patient, he had spastic paraparesis of 4/5 muscle strength in the extremities. There was a sensory defect up to the 12th thoracic vertebra. His deep tendon reflexes were hypoactive. The bilateral Babinski reflex was positive. He did not have urinary/fecal incontinence. His laboratory tests were normal. Contrast-enhanced spinal Magnetic Resonance Imaging (MRI) revealed a mass lesion originating from the 10th thoracic vertebral corpus, with a soft tissue component reaching approximately 12 mm in thickness in the paravertebral areas and an epidural component with a superior inferior distance of 35 mm in the spinal canal (Figure 1A & 1B).
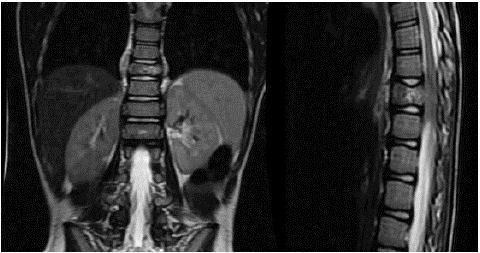
Figure 1A & 1B: Diagnostic magnetic resonance image of Case 1.
The patient was operated on by a neurosurgeon. The mass was partially excised by laminectomy at the 10th thoracic level and bilaterally at the 9th-11th thoracic level. According to the pathology result, CD99 and FLI-1 were positive, while SMA, desmin, and WT-1 were negative, and the case was referred to our clinic with a diagnosis of Ewing sarcoma. He did not have metastatic disease in whole-body scans performed with positron emission tomography using fluorine F-18-fludeoxyglucose (18F-FDG PET). After the operation, the PIAV treatment protocol consisting of ifosfamide, vincristine, cisplatin, and etoposide was initiated. After two cycles of chemotherapy, proton therapy was applied to the T9-11 level at a total dose of 45 Gray and a boost dose of 9 Gray. He completed the third cycle of chemotherapy with proton therapy. After the end of the treatment, the patient was followed up in remission for four years.
Case 2
One year after the diagnosis of the first case, his brother, aged 13 years, presented with complaints of swelling and pain in the right shoulder that developed after trauma and continued for two months. On his physical examination upon arrival, there was a palpable mass of 3*4 cm in diameter at the acromioclavicular junction of the right shoulder. His laboratory examinations were normal. The MRI of the shoulder revealed a bone lesion with exophytic growth toward the anterior at the distal end of the right clavicle. The lesion was associated with the medulla, and the continuity of the cortex was disrupted at the level of the lesion. The lesion with a lobulated contour had a maximum anteroposterior dimension of 33 mm, a vertical dimension of 18 mm, and a horizontal dimension of 49 mm. The lesion had a homogeneous internal structure and was hypointense near the muscle on T1-weighted images and hyperintense on T2-weighted images (Figure 2A & 2B).
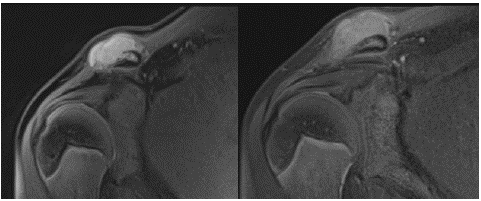
Figure 2A & 2B: Diagnostic magnetic resonance image of Case 2.
According to the results of the Tru-cut biopsy, the patient was diagnosed with Ewing sarcoma, with CD99, S-100, NSE, CD57, and FLI-1 focal positivity and desmin and myogenin negativity. Whole-body scans performed with 18F-FDG PET did not show metastatic disease. After he received two cycles of the PIAV protocol as had been applied to his brother, the tumor was excised with wide surgical margins by the orthopedics department. Bone tissue was irradiated during the operation. After the operation, the chemotherapy protocol was completed, and the treatment was terminated. He has been followed up in remission for the last three years.
Discussion
Ewing sarcoma was first described by James Ewing in 1921 [13]. To date, it has been reported in six sibling cases [14-16]. All these cases were girls, and four had metastatic disease (Table 1). Three cases died due to metastatic disease. Two of the remaining cases were in remission in their second year, while the remission period of the last case was not reported. Our patients are the first sibling cases reported in the literature. They did not have metastatic disease at diagnosis, and one has been followed up in remission for three years and the other for four years. Ewing sarcoma has been shown to have a better prognosis in girls [17]. However, the prognosis of our cases seems to be better than sister siblings described in the literature. It is difficult to make a further comment due to the absence of other brother sibling cases of Ewing sarcoma in the literature.
![]()
Report
Gender
Age, years
Involvement area
Metastatic disease
Outcome
Huntington et al. [14]
Girl
17
Tibia
Present
Mortality
Girl
18
Radius
Present
Mortality
Hutter et al. [15]
Girl
3
Rib
Present
Mortality
Girl
16
Metatarsus
Absent
In remission for two years
Joyce et al. [16]
Girl
9
Femur
Absent
In remission for two years
Girl
19
Thigh
Present
Remission period not specified
Table 1: Sibling cases with Ewing sarcoma reported in the literature.
In epidemiological studies on Ewing sarcoma, common factors have been found to be the family engaging in farming, older parental age, smoking habits of parents, a history of umbilical and inguinal hernias, and heart diseases in parents [18,19]. Our patients’ parents were not old and did not engage in farming. There was also no history of hernias. However, the mother had a history of an ablation procedure due to arrhythmia, and the father smoked at home.
Ewing sarcoma is not included in familial cancer syndromes, but it has been reported to be accompanied by some mutations (STAG2, TP53, and Rb1 genes) [20]. Due to the limited laboratory facilities of our hospital, genetic and molecular genetic studies could not be performed on the patients. According to the family history, the grandparents had been diagnosed with cancer but at an older age. There was no remarkable medical history in any other individual in the family at a young age. Therefore, we did not consider the possibility of familial cancer.
Despite advances in treatment, Ewing sarcoma is a highly aggressive tumor. Survival depends on the presence of metastases and tumor volume at the time of diagnosis [21]. A tumor diameter greater than 200 ml has been accepted as a poor prognostic factor [22,23]. Age is an important prognostic factor. Survival rates are lower in the adolescence period [24]. Neither of our patients had metastatic disease at the time of diagnosis, and their tumor volumes were less than 200 ml. The first case has been followed up in remission for four years and the second case for three years.
Approximately 2% of all Ewing sarcoma cases can develop into secondary malignancies. Secondary cases have worse survival than primary cases [25]. Neither of our cases had received prior chemotherapy or radiotherapy.
Hematological malignancies and solid tumors are seen in cases of Fanconi aplastic anemia. Patients with Ewing sarcoma and Fanconi mutations have been reported in the literature [26]. However, these patients have short stature, thrombocytopenia, and mild anemia. The height percentiles of our cases were normal, and there was no abnormality in their blood tests.
Conclusion
Our patients constitute the first male sibling cases of Ewing sarcoma reported in the literature. The inability to perform genetic and molecular genetic analyses can be considered a limitation of our case report. We consider that such an evaluation can be undertaken in future studies.
References
- Gurney JG, Swensen AR, Bulterys M. Malignant bone tumors. Cancer Incid Surviv Child Adolesc US SEER Program. 1975-1995: 99-110.
- Paulussen M, Craft AW, Lewis I, Hackshaw A, Douglas C, Dunst J, et al. Results of the EICESS-92 Study: two randomized trials of Ewing’s sarcoma treatment—cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol. 2008; 26: 4385-4393.
- Eaton BR, Claude L, Indelicato DJ, Vatner R, Yeh B, Schwarz R, et al. Ewing sarcoma. Pediatr Blood Cancer. 2021; 68: e28355.
- Esiashvili N, Goodman M, Marcus RB. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol. 2008; 30: 425-430.
- Valery PC, Holly EA, Sleigh AC, Williams G, Kreiger N, Bain C. Hernias and Ewing’s sarcoma family of tumours: a pooled analysis and meta-analysis. Lancet Oncol. 2005; 6: 485-490.
- Randall RL, Lessnick SL, Jones KB, Gouw LG, Cummings JE, Cannon-Albright L, et al. Is there a predisposition gene for Ewing’s sarcoma? J Oncol. 2010; 2010: 397632.
- Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I, Volk C, et al. Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases and remarkable consistency of t (11; 22)(q24; q12). Cancer Genet Cytogenet. 1988; 32: 229-238.
- Mugneret F, Lizard S, Aurias A, Turc-Carel C. Chromosomes in Ewing’s sarcoma. II. Nonrandom additional changes, trisomy 8 and der (16) t (1; 16). Cancer Genet Cytogenet. 1988; 32: 239-245.
- Lessnick SL, Braun BS, Denny CT, May WA. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene. 1995; 10: 423-432.
- Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, Healey JH, et al. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J Clin Oncol. 2005; 23: 548-558.
- Kovar H, Auinger A, Jug G, Aryee D, Zoubek A, Salzer-Kuntschik M, et al. Narrow spectrum of infrequent p53 mutations and absence of MDM2 amplification in Ewing tumours. Oncogene. 1993; 8: 2683-2690.
- Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001; 20: 4621-4628.
- Ewing J. Diffuse endothelioma of bone. In: Proc NY Pathol Soc. 1921; 21: 17-24.
- Huntington Jr RW, Sheffel DJ, Iger M, Henkelmann C. Malignant bone tumors in siblings: Ewing’s tumor and an unusual tumor perhaps a variant of Ewing’s tumor. JBJS. 1960; 42: 1065-1075.
- Hutter RV, Francis KC, Foote Jr FW. Ewing’s sarcoma in siblings: report of the second known occurrence. Am J Surg. 1964; 107: 598-603.
- Joyce MJ, Harmon DC, Mankin HJ, Suit HD, Schiller AL, Truman JT. Ewing’s sarcoma in female siblings: A clinical report and review of the literature. Cancer. 1984; 53: 1959-1962.
- Bacci G, Longhi A, Ferrari S, Mercuri M, Versari M, Bertoni F. Prognostic factors in non-metastatic Ewing’s sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Acta Oncol. 2006; 45: 469-475.
- Gargallo P, Yáñez Y, Juan A, Segura V, Balaguer J, Torres B, et al. Ewing sarcoma predisposition. Pathol Oncol Res. 2020; 26: 2057-2066.
- Johnson KJ, Carozza SE, Chow EJ, Fox EE, Horel S, McLaughlin CC, et al. Parental age and risk of childhood cancer: a pooled analysis. Epidemiol Camb Mass. 2009; 20: 475-83.
- Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014; 10: e1004475.
- Granowetter L, Womer R, Devidas M, Krailo M, Wang C, Bernstein M, et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children’s Oncology Group Study. J Clin Oncol. 2009; 27: 2536-41.
- Rodríguez-Galindo C, Liu T, Krasin MJ, Wu J, Billups CA, Daw NC, et al. Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children’s Research Hospital studies. Cancer. 2007; 110: 375-384.
- Karski EE, McIlvaine E, Segal MR, Krailo M, Grier HE, Granowetter L, et al. Identification of discrete prognostic groups in Ewing sarcoma. Pediatr Blood Cancer. 2016; 63: 47-53.
- Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HW, et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012; 30: 4148.
- Applebaum MA, Goldsby R, Neuhaus J, DuBois SG. Clinical features and outcomes in patients with secondary Ewing sarcoma. Pediatr Blood Cancer. 2013; 60: 611-615.
- Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016; 2: 616-624.