
Mini Review
J Ophthalmol & Vis Sci. 2016; 1(1): 1002.
Cone Rod Dystrophy in SCA1: Case Report and Review of Literature
Rashmi V1,2*
¹Neuro-Ophthalmology, W K Kellogg Eye Center, University of Michigan, USA
²Department of Neuro Ophthalmology, Shroff Eye Center, India
*Corresponding author: Verma Rashmi, Department of Neuro Ophthalmology, Shroff Eye Center, New Delhi, India
Received: January 24, 2016; Accepted: February 15, 2016; Published: February 22, 2016
Abstract
The Spinocerebellar Ataxias (SCAs) are a genetically and clinically diverse group of autosomal dominant disorders (labeled SCA1 through 36). Ophthalmic manifestations have been reported with many of them, but cone rod dystrophy has been reported predominantly with SCA7. Only 2 isolated cases in the US and recently members of a French family have been reported to have cone rod dysfunction with SCA1.We report hereby, an additional case of cone-rod dystrophy in a member of a family with genetically identified SCA1 in the United States.
Our patient presented with bilateral painless progressive vision loss with gait and balance difficulty. Family history was positive for ataxia in brother, father, 2 cousins and paternal aunt. Detailed clinical and electrophysiological studies including HVF, Multifocal ERG, Full field ERG and VEP were carried out. A diagnosis of cone rod dystrophy was made. The MRI was normal for age with no cerebellar or brain stem atrophy. Records of his brother, which showed positive genetic testing for SCA1, were retrieved and included after permission from him. The patient’s genetic testing further revealed SCA1. To our knowledge from a thorough Pubmed search, only 2 isolated cases in the US and recently members of a French family have been diagnosed to have cone rod dystrophy associated with SCA1. A complete neuro-ophthalmic exam including dilated fundus exam and electrophysiological testing in patients affected with unexplained visual loss in SCA1 might help in early diagnosis of cone rod dystrophy and in reporting more of such associations.
Keywords: SCA1; Ataxia; Ophthalmic manifestations; Cone rod dystrophy; Maculopathy
Background
The Spinocerebellar Ataxias (SCA’s) are a genetically and clinically diverse group of Autosomal Dominant (AD) disorders characterized by a slowly progressive cerebellar syndrome in association with various oculomotor, retinal, pyramidal, extra-pyramidal, sensory and cognitive/behavioral symptoms that vary with the underlying affected gene. The SCA’s are classified according to the genetic loci of the tri-nucleotide repeat and have been numbered according to their chronology of identification: SCA1 through SCA36. SCA 1, 2, 3, 6 and 7 comprise 80% of the SCA’s. They typically present in middle age, with slowly progressive in coordination of gait, often associated with appendicular ataxia, dysmetria, intentional tremors, gait difficulties, dysarthria and visual problems (difficulty focusing, diplopia, slowed/ dysmetric saccades). Atrophy of the cerebellum occurs frequently, causing the person to retain full mental capacity but progressively and irreversibly lose control of all motor functions. Brain imaging, in the form of MRI, helps rule out other diseases and localize the abnormality, usually atrophy of the cerebellum and brainstem. Recognition and discrimination of SCA’s are important for proper diagnosis and evaluation.
The common underlying mutational mechanism in all SCA’s is expanded CAG repeat encoding a tract of glutamine amino acids (Polyglutamine or Poly-Q tract) at different gene loci. In this inherited autosomal dominant disorder, the Poly-Q tract is increased from a normal level (maximum 35-40 repeat units) to a disease causing level and passed on to approximately 50% of the offspring. The age of onset varies inversely with length of Poly-Q tract repeats. The SCA’s exhibit genetic anticipation wherein the polyglutamine expansion lengthens when passed down to the progeny, resulting in earlier age of onset and more severe disease phenotype for individuals who inherit the disease allele.
SCA1
SCA1 was the first dominantly-inherited ataxia for which the locus and gene defect were identified [1]. Manifestations of SCA1 include severe four limb ataxia with dysarthria, causing most patients to be wheelchair-bound within 15-20 years of onset of disease. SCA1 is caused by poly-Q encoding CAG repeat expansions (39-83 repeats; normal alleles have 6-38 repeats) resulting in the SCA1 disease protein, Ataxin-1 mapped to chromosome 6p23) to have an abnormally long stretch of the amino acid glutamine and misfolded, resulting in neuronal degeneration and dysfunction in the cerebellum, brainstem and spinocerebellar tracts [2,3]. Ophthalmic manifestations include saccadic dysmetria in the form of hypermetric or slow saccades, gazeevoked and rebound nystagmus and reduced smooth pursuit gain [4]. Optic atrophy and consequently abnormal visual evoked potentials have been described in SCA1 [5]. Recently, 2 cases in the United States and members of a family in France have been reported to have cone rod dysfunction with SCA1 [6-8].
SCA7
SCA7 is relatively rare, autosomal dominant and the poly-Q encoding CAG repeat expansions (Greater than 36 repeats in disease, less than 20 repeats in health) occur within the ATXN7 gene mapped to chromosome 3p12-13 manifesting as gait imbalance, progressing to appendicular ataxia, and eventually leading to wheelchair confinement. Until recently, SCA7 was the only inherited ataxia reportedly associated with a cone-rod dystrophy manifesting as reduced visual acuity, dyschromatopsia, and photosensitivity, followed by progressive loss in peripheral vision and night blindness [9,10]. The ophthalmoscopic exam may initially have a normalappearing macula, progressing to retinal pigment epithelial mottling, eventually developing into a ‘‘bull’s-eye’’ maculopathy. However, Electroretinogram (ERG) demonstrating loss of photopic function or Ocular Coherence Tomography (OCT) showing thinning of the macula and Retinal Nerve Fiber Layer (RNFL) may help in diagnosing the macular and retinal dysfunction prior to the appearance of the maculopathy [11,12].
Methods
The patient, a 60 year old male, presenting with painless progressive bilateral vision loss, underwent clinical assessment including a dilated fundus exam, Humphrey Visual Field (HVF), fundus color and auto fluorescence photography, OCT maculae, VEP, Multifocal Electroretinography (multifocal ERG) and full-field ERG. He also underwent a brain MRI and a complete neurologic exam. Details of previous genetic examinations of his brother were extracted from medical charts after prior permission. After informed consent, DNA from the patient was analyzed for SCA1 gene mutation.
Results
The patient reported painless progressive bilateral vision loss for the past 2 years. Ocular history was significant for failed vision screening test at work 2 years earlier, which led to uneventful cataract surgery and subsequent YAG laser capsulotomy both eyes. Vision however failed to improve with actual further worsening of vision both eyes. Past medical history was significant only for hypertension, well controlled with single medication. However, in depth history revealed that he had balance difficulty for several years. His family history was positive for worsening ataxia in his brother, father (died at age 80), 2 cousins and a paternal aunt. His brother had tested positive for SCA1 in the past. His neurologic exam revealed normal strength and tone in all extremities, normal sensation and 2+ reflexes. However, he was found to manifest mild parkinsonian symptoms with a parkinsonian gait and postural instability.
Best-corrected visual acuity at distance was 20/60 in both eyes (OU). Near vision was J7 OU with a +3.00D reading add. Farnsworth D-15 was abnormal OU, consistent with congenital protanomalia. He was orthophoric in all directions of gaze; ocular motility was full but showed saccadic dysmetria. Pupils were equal, round and reactive with no afferent pupillary defect. Intraocular pressure by applanation was 16 mm Hg both eyes. Slit lamp exam of anterior segment revealed well centered posterior chamber implants and posterior vitreous detachments in both eyes. HVF 24-2 showed central scotomas both eyes (Figure 1). The dilated fundus exam revealed mild temporal pallor of both optic discs and subtle speckled macular pigmentary changes in both eyes (Figure 2 & 3). The multifocal ERG demonstrated a loss of P1 amplitude in the central 10 degrees in each eye (Figure 4A & 4B). Full field ERG showed relatively greater loss of cone-mediated responses compared to rod-mediated responses (Figure 5). The OCT of maculae revealed foveal atrophy and foveal cavitation, more in the right eye (Figure 6A & 6B). VEP latencies were essentially normal except for the delays at the small 7 minute checks.
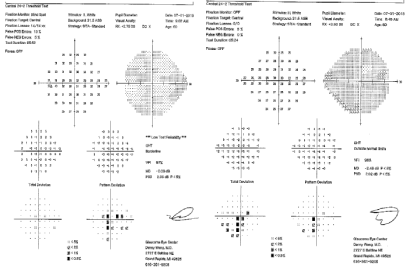
Figure 1: HVF (24-2) of the patient showing central scotomas both eyes.
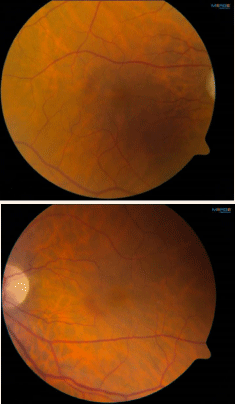
Figure 2: Fundus photos showing mild temporal pallor of both optic nerves
and subtle speckled macular pigmentary changes both eyes.
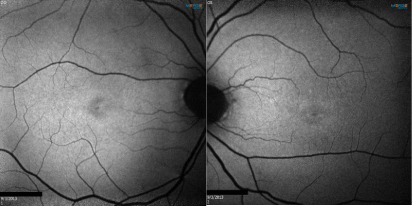
Figure 3: Fundus auto fluorescence showing macular pigmentary changes
both eyes.

Figure 4A: Multifocal ERG demonstrated a loss of P1 amplitude in the
central 10 degrees in each eye.
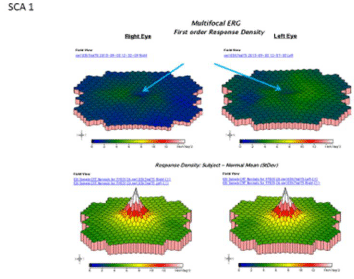
Figure 4B: Schematic color map of the multifocal ERG demonstrated a loss
of P1 amplitude in the central 10 degrees in each eye.
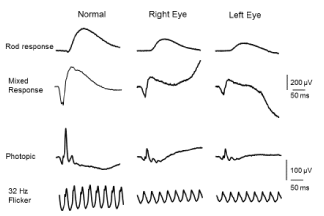
Figure 5: Full field ERG showed relatively greater loss of cone mediated
responses compared to rod mediated responses (cone/rod type of retinal
dysfunction).
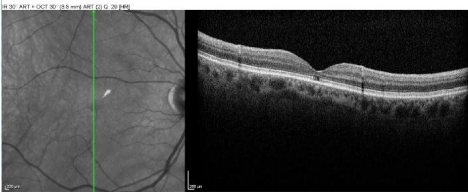
Figure 6A: The OCT of macula (OD) revealed foveal atrophy and foveal
cavitation.
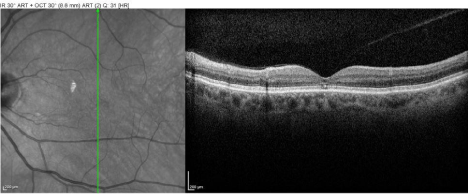
Figure 6B: The OCT of macula (OS) revealed minimal foveal atrophy and
foveal cavitation.
He was found to have a normal MRI brain for age with no cerebellar or brain stem atrophy (Figure 7). The diagnosis of SCA1 was confirmed via molecular analysis with a 42 CAG repeat expansion in one allele. Records of the patient’s brother revealed similar visual complaints and positive genetic testing for SCA1.
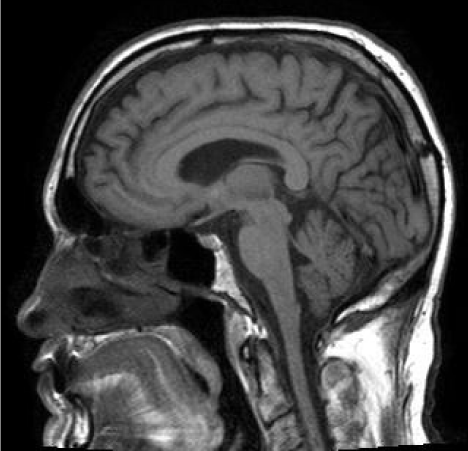
Figure 7: Normal MRI brain for age with no cerebellar or brain stem atrophy.
Discussion
Retinal changes in autosomal dominant SCAs have been reported with the spectrum ranging from attenuated retinal arteries, mild temporal disc pallor, normal fundus auto fluorescence to severe visual loss, with cone rod dystrophy being reported until recently only with SCA7 [13-15]. However, the most common finding on fundus exam is a maculopathy/cone-rod dystrophy, beginning with retinal pigment epithelial mottling in the macular area, finally developing into a ‘‘bull’s-eye’’ maculopathy. Multifocal ERG may be more sensitive than full-field ERG in early stages of the disease [16].
SCA1 has previously been reported to be associated only with optic neuropathy as a cause of visual impairment [17]. However, Stricker et al., used OCT to demonstrate selective involvement of the temporal retinal nerve fiber layer, built primarily by the papillomacular bundle and attributed it to a differential vulnerability of these retinal axons to mutated ataxin1 [18]. Pula et al., reported significant thinning of macular volume in seven SCA1 patients and Vaclavik et al. described an SCA1 patient with maculopathy and retinal dysfunction [19,20].
An extensive PubMed search has revealed only 2 case reports of isolated patients and another study of members of a single French family of frank maculopathy/cone-rod dystrophy associated with SCA1. Saito et al., described a patient with unexplained progressive loss of vision and a pigmentary macular dystrophy similar to that seen in SCA7. Full-field ERG revealed photoreceptor dysfunction and genetic testing had negative results for SCA7, but positive for SCA1, suggesting an association between macular dystrophy and SCA1 [6]. Thurtell et al., have described a case of a genetically proven SCA1 patient with rod cone dystrophy presenting with unexplained visual loss. Fundus exam was suggestive of subtle pigmentary macular degeneration but no optic atrophy. Full-field ERG revealed cone rod dysfunction [7]. Lebranchu et al., recently reported a crosssectional clinical and electrophysiological study of four members of a French family with unexplained visual loss and genetically confirmed SCA1with fundus picture of subtle maculopathy [8]. OCT demonstrated either central thinning of the retina or disorganized photoreceptor layers in all patients. Multifocal ERG revealed central retinal dysfunction in one patient. They also discussed the very low probability of an SCA1 family exhibiting a hereditary macular disease and speculated both diseases to be genetically linked, either with two independent mutations on the same chromosome or because of a “local effect” of one mutation another gene nearby [8].
Our study reports another patient, member of a family with genetically identified SCA1in the United States, affected by central visual loss related to cone-rod dysfunction and puts forth the possibility of more of these associations being diagnosed and reported in the future.
Is it that these are new associations or that these findings have previously not been actively looked for or documented? The patients with SCA7 develop retinal findings before the onset of ataxia and so are brought to attention sooner; however, it is possible that the patients with SCA1 develop the ataxia before any ophthalmic/retinal findings and are incapacitated or wheelchair bound (ataxia is more severe and of early onset in SCA1 as compared to SCA7) [3,16]. These patients are either not aware of or do not complain of the mild to moderate decline in visual function sooner!
Confirmation of the SCA1 mutation with ataxia and similar visual symptoms in at least one other family member of our patient consolidates our findings and diagnosis and suggests that the ophthalmic manifestations in these patients are part of the disease and not mere incidental findings. Other family members of the patient declined evaluation either because they did not suffer from visual loss and did not desire genetic testing or were too incapacitated or wheelchair bound for easy transportation and evaluation. Patients with SCA1 should be evaluated by multifocal ERG and other electrophysiological tests at the earliest development of unexplained vision loss with the abovementioned findings in mind.
Conclusion
Cone-rod dysfunction/maculopathy has sparingly been reported to be associated with SCA1. A complete neuro ophthalmic exam including dilated fundus exam can help in actively looking for these findings in patients with SCA1 and unexplained vision loss. A possible explanation that these have not been reported often with SCA1 might be that patients with severe disease might have overwhelming motor manifestations and the visual disturbance might be under reported by the patient. Early and prompt ophthalmic screening of SCA1 patients with particular attention to maculopathy/cone-rod dystrophy would help bring to light more such cases.
References
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007; 30: 575-621.
- Kang S, Hong S. Molecular pathogenesis of spinocerebellar ataxia type 1 disease. Mol Cells. 2009; 27: 621-627.
- Paulson HL. The spinocerebellar ataxias. J Neuroophthalmol. 2009; 29: 227- 237.
- Buttner N, Geschwind D, Jen JC, Perlman S, Pulst SM, Baloh RW. Oculomotor phenotypes in autosomal dominant ataxias. Arch Neurol. 1998; 55: 1353-1357.
- Lee WY, Jin DK, Oh MR, Lee JE, Song SM, Lee EA, et al. Frequency Analysis and Clinical Characterization of Spinocerebellar Ataxia Types 1, 2, 3, 6, and 7 in Korean Patients. Arch Neurol. 2003; 60: 858-863.
- Saito Y, Matsumura K, Shimizu T, Ichikawa Y, Ochiai K, Goto J, et al. Pigmentary Macular Dystrophy in Spinocerebellar Ataxia Type 1. J Neurol Neurosurg Psychiatry. 2006; 77: 1293.
- Thurtell MJ, Biousse V, Newman NJ. Rod-cone dystrophy in spinocerebellar ataxia type 1. Arch Ophthalmol. 2011; 129: 956-958.
- Lebranchu P, Le Meur G, Magot A, David A, Verny C, Weber M, Milea D. Maculopathy and spinocerebellar ataxia type 1: a new association? J Neuroophthalmol. 2013; 33: 225-231.
- Hamel CP. Cone rod dystrophies. Orphanet J Rare Dis. 2007; 2: 7.
- Garden G. Spinocerebellar Ataxia Type 7. Pagon RA, Adam MP, Arlinder HH, Wallace SE, Amemiya A, Bean LJH, et al. editor In: GeneReviews. University of Washington, Seattle, 1993.
- Hugosson T, Granse L, Ponjavic V, Andréasson S. Macular dysfunction and morphology in spinocerebellar ataxia type 7 (SCA 7). Ophthalmic Genet. 2009; 30: 1-6.
- Ahn JK, Seo JM, Chung H, Yu HG. Anatomical and Functional Characteristics in Atrophic Maculopathy Associated with Spinocerebellar Ataxia Type 7. Am J Ophthalmol. 2005; 139: 923-925.
- Marelli C, Cazeneuve C, Brice A, Stevanin G, Dürr A. Autosomal dominant cerebellar ataxias. Rev Neurol. 2011; 167: 385-400.
- Gouw LG, Digre KB, Harris CP, Haines JH, Ptacek LJ. Autosomal Dominant Cerebellar Ataxia with Retinal Degeneration: Clinical, Neuropathologic, and Genetic Analysis of Large Kindred. Neurology. 1994; 44: 1441-1447.
- Thurtell MJ, Fraser JA, Bala E, Tomsak, RL, Biousse V, Leigh RJ, et al. Two Patients with Spinocerebellar Ataxia Type 7 Presenting with Profound Binocular Visual Loss yet Minimal Ophthalmoscopic Findings. J Neuroophthalmol. 2003; 29: 187-191.
- Miller RC, Tewari A, Miller JA, Garbern J, Van Stavern GP. Neuroophthalmologic features of spinocerebellar ataxia type 7. J Neuroophthalmol. 2009; 29: 180-186.
- Abe T, Abe K, Aoki M, Itoyama Y, Tamai M. Ocular changes in patients with spinocerebellar degeneration and repeated trinucleotide expansion of spinocerebellar ataxia type 1 gene. Arch Ophthalmol. 1997; 115: 231-236.
- Stricker S, Oberwahrenbrock T, Zimmermann H, Schroeter J, Endres M, Brandt AU, et al. Temporal retinal nerve fiber loss in patients with spinocerebellar ataxia type 1. PLoS One. 2011; 6: e23024.
- Pula JH, Towle VL, Staszak VM, Cao D, Bernard JT, Gomez CM. Retinal Nerve Fibre Layer and Macular Thinning in Spinocerebellar Ataxia and Cerebellar Multisystem Atrophy. Neuroophthalmology. 2011; 35: 108-114.
- Vaclavik V, Borruat FX, Ambresin A, Munier FL. Novel maculopathy in patients with spinocerebellar ataxia type 1 autofluorescence findings and functional characteristics. JAMA Ophthalmol. 2013; 131: 536-538.