
Research Article
J Ophthalmol & Vis Sci. 2016; 1(1): 1005.
Characterization of the Basal Proximal Promoter of the Human Retinol Dehydrogenase 10 Gene
Farjo KM* and Ma JX
Department of Physiology, University of Oklahoma Health Sciences Center, USA
*Corresponding author: Krysten M Farjo, Department of Physiology, University of Oklahoma Health Sciences Center, USA
Received: April 15, 2016; Accepted: May 24, 2016; Published: May 26, 2016
Abstract
Retinol Dehydrogenase 10 (RDH10) is essential for retinoic acid synthesis during embryonic development, and previous studies have shown that RDH10 can also catalyze the final step of the retinoid visual cycle to generate the visual chromophore 11-cis-retinal. RDH10 is widely expressed during embryogenesis, but its expression becomes restricted in adulthood to the retinal Muller cells and Retinal Pigment Epithelium (RPE). Interestingly, RDH10 expression is very high in RPE, the cell type in which the retinoid visual cycle generates 11-cisretinal. Despite the importance of RDH10 in embryonic retinoic acid synthesis, and the high probability that RDH10 also contributes to the generation of visual chromophore, very little is known about how RDH10 expression is regulated. Previous studies have shown that RDH10 mRNA expression correlates with protein expression, suggesting that RDH10 expression is primarily controlled at the level of transcriptional regulation. The present study utilizes luciferase reporter assays and electrophoretic mobility shift assays to characterize the basal proximal promoter of the human RDH10 gene, which spans up to -923 base pairs before the transcription start site. We also identify a specific transcription factor II B (TFIIB) binding site that is necessary for maximum promoter activity in human Telomerase-immortalized RPE (hTERT-RPE1) cells. TFIIB is important for recruiting the RNA polymerase II complex to initiate transcription, but it can also aid in the recruitment of tissue-specific transcription factors that selectively amplify gene expression. Thus, identification of a pertinent TFIIB site in the RDH10 proximal promoter is important for understanding the transcriptional regulation of RDH10 gene expression to produce widespread expression in embryonic tissues and later maintain and amplify expression in adult RPE.
Keywords: Retinol dehydrogenase; Retinoid visual cycle; Retinoic acid; Retinal pigment epithelium; Retinal Muller cells; Transcriptional regulation
Abbreviations
RDH10: Retinol Dehydrogenase 10; RPE: Retinal Pigment Epithelium; TFIIB: Transcription Factor II B; hTERT-RPE1: human Teleomerase-immortalized RPE cells; atRA: all-trans Retinoic Acid; 11cRAL: 11-cis-Retinal; RARs: nuclear Retinoic Acid Receptors; atROL: all-trans-Retinol; atRAL: all-trans-Retinal; 11cROL: 11-cis-retinol; TSS: Transcription Start Site; bp: base pairs; 3’UTR: 3’Untranslated Region; RNA pol II: RNA polymerase II complex; EMSA: Electrophoretic Mobility Shift Assays; CREB: Cyclic-AMP response element
Introduction
Vitamin A is an essential nutrient that is metabolized to form its biologically-active derivatives, most notably all-trans Retinoic Acid (atRA) and 11-cis-retinal (11cRAL). atRA is a signaling molecule that binds to nuclear Retinoic Acid Receptors (RARs) to modulate gene expression, and atRA-mediated gene regulation drives several aspects of embryonic development and is necessary for immune function, skin and bone health, and reproduction in adults [1,2]. 11cRAL is generated specifically in the Retinal Pigment Epithelium (RPE), and serves as the photosensitive chromophore that is essential for visual transduction in the retina [3,4]. Defective metabolism of Vitamin A can cause birth defects, decreased reproductive capacity, immune system deficiencies, skin diseases, and visual deficiencies, including blindness [1,2,4].
Vitamin A metabolism is controlled by regulating the spatiotemporal expression of Vitamin A-metabolizing enzymes that catalyze distinct steps in the metabolism of Vitamin A to atRA and 11cRAL. The biosynthesis of atRA requires two sequential oxidative reactions. First, Vitamin A, all-trans-Retinol (atROL), is oxidized to form all-trans-Retinal (atRAL). Then atRAL is oxidized to generate atRA. Our previous studies have shown that Retinol Dehydrogenase 10 (RDH10) is essential during embryonic development to catalyze the first step of atRA synthesis, the oxidation of all-trans-Retinol (atROL) to all-trans-Retinal (atRAL) [5,6]. RDH10 is widely expressed during mouse embryogenesis in a variety of developing organ systems, including the central nervous system, kidney, respiratory tract, cardiac system, digestive system, spinal column, forelimb buds, craniofacial structures (nose, eyes, ears, teeth), and lymphatic tissue [5,7,8]. RDH10 loss-of-function in mice causes embryonic lethality and abnormalities in nasal, optic, otic, forelimb, cardiac, lung, liver, gut, gonad, vascular, and neuronal development [5,6].
In the adult, RDH10 expression becomes restricted and is primarily limited to retinal Muller cells and the RPE, which expresses a large amount of RDH10 [9,10]. This is very interesting, since the RPE serves to generate the visual chromophore 11cRAL through a series of enzymatic reactions known as the “retinoid visual cycle” [11-13], suggesting that RDH10 expression may be maintained in the RPE and Muller cells because it contributes to the biosynthesis of 11cRAL. We have previously shown that RDH10 catalyzes the final step of 11cRAL synthesis, the oxidation of 11-cis-Retinol (11cROL) to 11cRAL, in vitro [14]. This suggests that RDH10 expression may be maintained in the RPE and Muller cells because it contributes to the biosynthesis of 11cRAL.
Thus, RDH10 is the only Vitamin-A metabolizing enzyme that has been implicated to function in both major pathways of Vitamin A metabolism.
Despite the clear physiological importance of RDH10, very little is known about how RDH10 expression is regulated. Previous studies have shown that RDH10 mRNA expression correlates with protein expression [9,10], suggesting that RDH10 is primarily regulated at the level of transcription. However, transcriptional elements in the promoter of RDH10 have not been identified. The human RDH10 gene is comprised of 6 exons spanning over 30 kb and is located on chromosome 8q21.11 (Genbank ID: AF456765). The in silico predicted Transcription Start Site (TSS) is 260 base pairs (bp) upstream of the translation start codon. However, a previous study found that a different TSS, located 688 bp upstream of the start codon, is utilized in the A549 lung cell line by 5’ primer extension analysis [15]. A putative TATA box lies 25 bp upstream of this empiricallyidentified TSS, while no TATA box is present near the in silicopredicted TSS. Two RDH10 mRNA transcripts of 3 kb and 4 kb have been detected in several human tissues by northern blotting [15]. No alternative splicing was detected by RT-PCR analysis, but two distinct polyadenylation sites are utilized in A549 cells, suggesting that the two RDH10 transcripts likely reflect the utilization of different polyadenylation sites [15]. The putative TSSs that are located at 260 bp and 688 bp upstream of the ATG start codon of RDH10 signifies that essential promoter elements may exist in the proximal promoter at a location near, but beyond 688 bp upstream of the ATG.
The present study defines the basal proximal promoter of the human RDH10 gene, including a putative transcription factor II B (TFIIB) binding site that is necessary for the highest level of basal transcription in human Telomerase-immortalized RPE cells (hTERTRPE1). TFIIB recruits the RNA polymerase II complex (RNA pol II) to initiate transcription [16-18], and TFIIB also has multiple binding domains that are involved in the recruitment of tissue-specific transcription factors that selectively amplify gene expression [19,20]. Thus, identification of a pertinent TFIIB site in the RDH10 proximal promoter is important for determining how RDH10 transcription is induced in specific embryonic tissues and maintained in adult RPE and retinal Muller cells.
Materials and Methods
Construction of vectors
The 5’flanking DNA of the human RDH10 gene was sequenced up to -1386 bp from the Genbank-predicted TSS and cloned into the pGL3-Basic luciferase reporter plasmid using KpnI and MluI restriction sites in the multiple cloning site of the pGL3-Basic vector.
The 5’-deletion constructs were created by various restriction enzyme digestions and subsequent re-ligations. Several constructs were created by first digesting the -1386 bp plasmid with KpnI to linearize the vector. Then the vector was subsequently digested with a second restriction endonuclease in order to remove the unwanted portion of the 5’end of the RDH10 promoter sequence. The second restriction endonucleases that were used for each construct are as follows: NsiI for -1209 bp, BstBI for -923 bp, PpuMI for -848 bp, NheI for -756 bp, SstI for -687 bp. All of the luciferase reporter constructs contain the same 3’end at +173 bp from the Genbank-predicted TSS.
Cell culture and transient DNA transfection
Human Telomerase-immortalized RPE cells (hTERT-RPE1) were purchased from American Type Culture Collection, (ATCC, Manassas, VA). The cells were cultured at 37oC in 5% CO2/95% air in DMEM supplemented with 10% FBS. Cells were transfected using FuGENE 6 according to the manufacturer’s protocol. Briefly, cells were grown to between 50% and 80% confluency. DMEM was removed, cells were rinsed in PBS and then OptiMEM was added to the cells for the duration of the transfection period. DNA was premixed with FuGENE 6 reagent at a DNA to FuGENE 6 ratio of 1:3, and then added to the cells. After 6 hrs of transfection, the OptiMEM/DNA/FuGENE 6 transfection mixture was removed and replaced with DMEM/10% FBS until the time of processing for the luciferase assay.
Luciferase reporter assays
The dual luciferase reporter assay system kit (Promega, Madison, WI) was utilized for luciferase reporter assays according to the manufacturer’s protocol. hTERT-RPE1 cells were seeded into 12- well plates at 65,000 cells per well, and allowed to attach and spread on the plate overnight. At time of transfection, cells were roughly 65% confluent. The pRL-TK vector, expressing renilla luciferase under control of the thymidine kinase promoter was transfected into all cells at 100 ng/well. The pGL3-Basic vector and the pGL3- RDH10 5’flanking sequence vectors, encoding firefly luciferase, were transfected into subsets of cells as indicated at 400 ng/well.
Approximately 30 h post-transfection, cells were harvested in 250 μl of passive lysis buffer/well (Promega, Madison, WI). The light generated by 20 μl of each cell lysate was analyzed using a Berthold luminometer (Berthold Technologies USA, Oak Ridge, TN) and the dual luciferase assay kit (Promega), which utilizes special reagents that allow the light units produced by firefly luciferase to be distinguished from light units produced by renilla luciferase. The light units emitted by firefly luciferase were divided by the light units emitted by renilla luciferase to normalize results according to differences in transfection efficiency.
Preparation of nuclear extracts
Nuclear extracts were prepared using an Active Motif kit (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol. Briefly, hTERT-RPE1 cells were harvested at 90% to 100% confluency by removing the cell media and rinsing and collecting the cells in ice-cold PBS including phosphatase inhibitors. Cells were briefly centrifuged at 500 x g and then resuspended in hypotonic lysis buffer containing 0.5% of a proprietary detergent (Active Motif) for 15 min on ice. Cells were centrifuged at 14,000 x g for 30 sec, and the supernatant (cytosolic extract) was removed. The pellet (nuclear fraction) was resuspended in a proprietary nuclear extraction buffer (Active Motif) containing 1 mM DTT and protease inhibitors, rocked for 30 min on ice, vortexed, and centrifuged at 14,000 x g for 10 min. The supernatant (nuclear extract) was frozen in liquid nitrogen and stored at -80oC. Protein concentration was determined by Bradford method [21], and the quality of nuclear extracts was evaluated by western blotting as described previously [14] with an anti-fibrillarin antibody.
Electrophoretic Mobility Shift Assays (EMSA)
The 167-bp double-stranded DNA probe was generated by digesting the pGL3-(-1386) plasmid with BstBI and NheI restriction endonucleases. The probe was purified by agarose gel electrophoresis and gel extraction. The probe was end-labeled using [γ-32P]ATP (3000 Ci/mmol, PerkinElmer, Waltham, MA) and T4 polynucleotide kinase (Promega, Madison, WI), and then purified using a G25 column. The binding reaction included nuclear extract, gel shift binding buffer (4% glycerol, 1 mM MgCl2, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris, pH 7.5), and 32P-labeled DNA probe in a total volume of 10 to 20 μl, depending on the experiment. Additional factors, such as unlabeled DNA probes, EDTA, poly-dI-dC, and ZnCl2, were added to theb binding reaction as indicated. The binding reaction was allowed to proceed for 20 minutes at RT, and then samples were electrophoresed on a 5% non-denaturing polyacrylamide gel with 0.5X TBE buffer (44.5 mM Tris, 44.5 M boric acid, 1 mM EGTA) at 250 V. Gels were dried under a vacuum for 45 min at 70°C, and then exposed to Kodak Biomax MR film at -80°C.
Results
Identification of the human RDH10 proximal promoter
To define the promoter of the human RDH10 gene, the genomic DNA sequence located 5’ of the RDH10 gene start codon was cloned into a luciferase reporter plasmid. The TSS (as indicated by Genbank ID: AF4567655) is defined as position +1. The cloned “full length” 5’-flanking sequence used in this study begins at -1386 bp upstream of the TSS and ends at +173 bp downstream of the TSS. Truncations of the full length 5’ flanking sequence were generated and tested, and each 5’flanking sequence is identified based on where the 5’ end of the sequence terminates in relation to the TSS. For instance, the “full length” sequence is referred to as “-1386 bp” (Figure 1). hTERT-RPE1 cells were transfected with luciferase reporter plasmids and luciferase activity was measured approximately 30 h post-transfection. The negative control plasmid pGL3-Basic, which has no 5’flanking sequence inserted, was used as a reference in all assays and is referred to simply as “Basic”. Luciferase activity was similar in all constructs tested between -280 bp and -756 bp (Figure 2). However, the -923 bp construct had a 5-fold induction in promoter activity compared to -756 bp, indicating that potent cis-regulatory elements are present in the sequence between -756 bp and -923 bp (Figure 2).
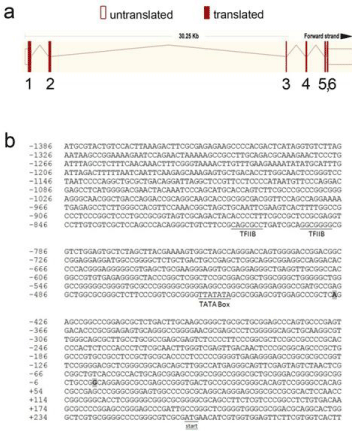
Figure 1: The human RDH10 gene structure and 5’ flanking DNA sequence.
a. Schematic diagram of theexon/intron structure of the human RDH10
gene is shown as adapted from www.ensembl.org. Shaded and open boxes
represent exons and UTRs, respectively. The connecting lines represent
introns. b. The 5’ flanking sequence of the human RDH10 gene is shown.
Numbers on the left indicate the nucleotide position relative to the Genbankpredicted
TSS, which is defined as position +1 and is shaded in gray. The
TSS identified in A549 cells by Picozzi et al. is also shaded in gray, and is
located -427 bp from the Genbank TSS. The ATG start codon is underlined
and labeled. The TFIIB sites predicted by the MatInspector program are
underlined and labeled, as is the predicted TATA box.
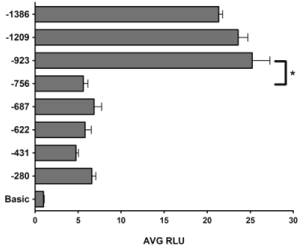
Figure 2: Promoter activity of the 5’ flanking DNA sequence of the RDH10
gene. Luciferase reporter plasmids were constructed using the pGL3-Basic
vector (Promega) and the 5’ flanking sequences of the human RDH10
gene. The position of the 5’end of each fragment is indicated relative to
the Genbank TSS at position +1. hTERT-RPE1 cells were transfected
with the indicated luciferase reporter plasmids, and luciferase activity was
measured approximately 30 h post-transfection. All values are compared to
the negative control plasmid, Basic. The data shown represent the Average
Relative Luciferase Units (AVG RLU) and standard deviation for more than
three independent experiments. *P<0.05 based on one-way ANOVA and
Bonferroni post-hoc test.
Nuclear protein(s) binds to a 167-bp sequence in the proximal promoter
In order to analyze the binding of nuclear transcription factors, the 167 bp sequence ranging from -756 bp to -923 bp was isolated to use as a probe in Electrophoretic Mobility Shift Assays (EMSA). Nuclear extracts were prepared from hTERT-RPE1 cells and the relative quality of nuclear extract preparations was confirmed by western blotting with an anti-fibrillarin (a nucleolar-specific protein) antibody (Figure 3a). Incubation of the 32P-labelled 167-bp probe with nuclear extract yielded one stable complex as indicated by retardation of the 167-bp probe migration (Figure 3b). This complex appeared to result from specific binding of nuclear protein to the probe, as this complex remained stable in the presence of non-specific competitor DNA, poly-dIdC (Figure 4a). This protein-DNA interaction was specific for the 167-bp sequence, as excess amount of unlabelled 167- bp probe competed away most of the complex formation (Figure 4b & 4c), while an excess amount of an unlabelled oligonucleotide containing a Cyclic-AMP Response Element (CREB)-binding site did not dissociate the complex (Figure 4c).
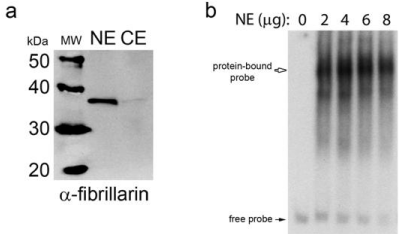
Figure 3: Nuclear protein binds to the 167-bp DNA promoter sequence.
a. Nuclear protein Extracts (NE) were prepared from hTERT-RPE1 cells and
analyzed by western blotting with an anti-fibrillarin antibody. Cytosolic Extract
(CE) was also analyzed. b. The 32P-labeled 167-bp DNA sequence probe
corresponding to the 5’-flanking sequence of the RDH10 gene ranging from
-756 bp to -923 bp was incubated with increasing amounts of hTERT-RPE1
nuclear extract, and then the binding reactions were analyzed by EMSA.
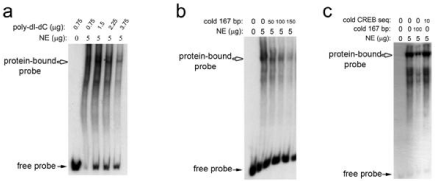
Figure 4: Nuclear protein forms a specific complex with the 167-bp DNA
promoter sequence.
a. EMSA demonstrates that the shift of the 32P-labeled 167-bp probe results
from a specific protein-DNA interaction, since a non-specific competitor DNA
fragment, poly-dI-dC, did not compete with the 167-bp probe and dissociate
the complex. b. Addition of 50-, 100-, and 150-fold molar excess unlabeled
167-bp DNA caused the complex to dissociate, demonstrating the unlabeled
167-bp DNA fragment competed for binding to nuclear protein with the
32P-labeled 167-bp DNA probe. c. An unlabelled oligonucleotide containing a
CREB-binding sequence also did not dissociate the complex.
A zinc-dendent transcription factor binds the 167-bp sequence
In silico analysis using the MatInspector program (www. genomatix.de) predicted at least 10 zinc-dependent transcription factor binding sites within the 167-bp probe based on a core matrix similarity of 100% and matrix similarity threshold of > 0.90 (data not shown). To determine if a zinc-dependent transcription factor binds to the 167-bp probe, all divalent cations were chelated by addition of 1 mM EDTA, which completely blocked the formation of the protein- DNA complex (Figure 5). Addition of 2 mM ZnCl2 restored the protein-DNA complex formation, indicating that a zinc-dependent transcription factor(s) binds to the 167-bp probe (Figure 5).
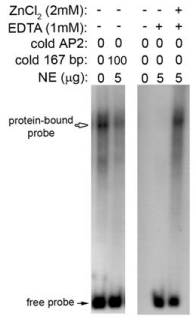
Figure 5: Binding of nuclear protein to the 167-bp DNA promoter sequence
is zinc-dependent. EMSA demonstrates that addition of EDTA completely
blocks formation of the nuclear protein-DNA complex. Addition of excess
ZnCl2 can restore complex formation in the presence of EDTA.
Identification of a 92-bp sequence containing putative TFIIB-binding elements
To narrow down the pool of candidate zinc-dependent transcription factor binding sites present in the 167-bp DNA sequence located between -756 bp and -923 bp upstream of the TSS, a luciferase reporter plasmid terminating at -48 bp upstream of the TSS was constructed. Luciferase assays demonstrated that the 92-bp sequence between -756 bp and -848 bp confers a 3-fold induction in promoter activity, while the 75-bp sequence between -848 bp and -923 bp resulted in less than a 2-fold induction (Figure 6). This indicates that the 92-bp sequence between -756 bp and -848 bp is more important for promoter activity, and likely contains the binding site for the zinc-dependent transcription factor(s). In silico analysis predicted two TFIIB binding elements separated by 8 bp within this 92-bp sequence (Figure 1b). TFIIB is an essential basal transcription factor that is necessary to recruit RNA pol II and initiate transcription [16- 18]. TFIIB contains a zinc ribbon domain that is essential for binding to and thereby recruiting RNA pol II [22-24]. Thus, these putative TFIIB binding elements were considered as the key sequences within the 92-bp fragment that may be responsible for the 3-fold induction in promoter activity of the -848 bp versus -756 bp construct.
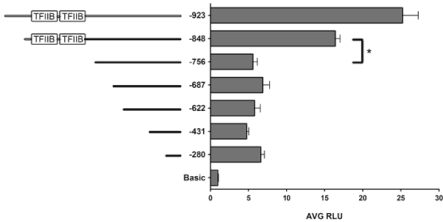
Figure 6: A 92-bp DNA promoter sequence, including 2 putative TFIIB
binding sites, confers a 3-fold induction in promoter activity. In an effort
to narrow down the pool of potential transcription factor binding sites that
could be responsible for the protein-DNA interaction observed by EMSA
with the 167-bp DNA probe, a new luciferase reporter plasmid, -848 bp, was
constructed. The relative lengths of each fragment are indicated by solid lines
to the left of the graph, and the relative locations of the putative TFIIB sites
are indicated. The data shown represent the Average Relative Luciferase
Units (AVG RLU) and standard deviation for more than three independent
experiments. *P<0.05 based on one-way ANOVA and Bonferroni post-hoc
test.
Mutation of TFIIB binding sites decreases promoter activity
To determine if the in silico-predicted TFIIB binding sequences are important for RDH10 promoter activity, three additional luciferase reporter plasmids were constructed, all of which spanned the -1386 bp sequence upstream of the TSS, but had targeted deletion of either the upstream TFIIB site, the downstream TFIIB site, or the sequence spanning both TFIIB sites. Luciferase reporter assays demonstrate that deletion of either the upstream TFIIB site or both TFIIB sites significantly reduces promoter activity (Figure 7). However, deletion of only the downstream TFIIB site did not decrease promoter activity (Figure 7). This suggests that the upstream TFIIB site is important for promoter activity, although deletion of this site results in less than a 2-fold reduction in promoter activity, indicating that this site is not absolutely essential for promoter activity.
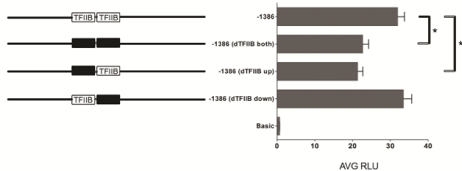
Figure 7: The human RDH10 gene structure and 5’ flanking sequence. To
determine the importance of the predicted TFIIB sites for promoter activity,
each TFIIB site was deleted individually or together in the -1386 bp luciferase
reporter plasmid. The relative lengths of each fragment are indicated by
solid lines to the left of the graph, and the relative locations of the putative
TFIIB sites are indicated. Black boxes cover the TFIIB sites that are deleted
in the plasmids. The data shown represent the Average Relative Luciferase
Units (AVG RLU) and standard deviation for more than three independent
experiments. *P<0.05 based on unpaired t-test.
Discussion
In the present study, we characterized the proximal promoter sequence of the RDH10 gene that is necessary for maximal promoter activity in hTERT-RPE1 using luciferase reporter assays. Through promoter deletion analyses, we identify a specific promoter sequence of only 92-bp that is necessary for maximum transcription. Using EMSA, we demonstrate that transcription factor binding to the 92- bp sequence is zinc-dependent. Site-specific deletion of two predicted TFIIB-binding sites identifies a single TFIIB site located at -810 bp from the TSS that is necessary to maintain the highest level of basal transcription from the RDH10 proximal promoter. TFIIB is a crucial basal transcription factor that is necessary to recruit RNA pol II to initiate transcription [16-18], but TFIIB also has multiple binding domains that are involved in the recruitment of tissue-specific transcription factors that selectively amplify gene expression [19-20]. Thus, identification of a pertinent TFIIB site in the RDH10 proximal promoter will lay the foundation for future studies to understand the transcriptional regulatory mechanisms that control RDH10 gene expression during embryonic development and also maintain and amplify expression in adult RPE.
The longest RDH10 proximal promoter sequence tested was -1386 bp from the Genbank-predicted TSS, and this sequence provided more than a 20-fold induction in promoter activity over the empty pGL3-Basic plasmid. However, the -923 bp sequence had the same level of promoter activity as the -1386 bp sequence (Figure 2), indicating that the -923 bp sequence encompasses the basal promoter sequence of the RDH10 gene. Truncation to -756 bp upstream of the TSS reduced promoter activity by 5-fold, signifying that the 167-bp sequence between -756 bp and -923 bp is essential for basal promoter activity. EMSA demonstrated that the 167-bp sequence binds specifically to nuclear protein, and that formation of a protein-DNA complex is zinc-dependent. Eleven zinc-dependent transcription factor binding sites are predicted within the 167-bp sequence using the MatInspector program. To reduce the number of candidate zincdependent transcription factors responsible for binding to the 167-bp sequence, we generated and tested an additional luciferase reporter construct, -848 bp. The -848 bp sequence conferred a 3-fold increase in promoter activity over the -756 bp construct, which signifies that the most crucial transcription factor binding sites are present in the 92-bp sequence between -756 bp and -848 bp. The MatInspector program predicted only six zinc-dependent transcription factor binding sites, including two TFIIB binding sites, within this 92-bp sequence. TFIIB is a basal transcription factor that is loosely defined as a zinc-dependent transcription factor, because it does not strictly require zinc for binding to DNA, although the zinc ribbon domain of TFIIB is necessary for TFIIB to bind and recruit RNA pol II.
Since TFIIB binding to DNA is necessary to recruit RNA pol II, we focused on determining if the predicted TFIIB-binding sites were important for RDH10 promoter activity. The luciferase reporter plasmid containing the -1386 bp sequence was modified to generate sequences lacking either the upstream TFIIB site, the downstream TFIIB site, or both sites. These studies demonstrate that the upstream TFIIB site is necessary for maximum basal transcription, while deletion of the downstream TFIIB site alone does not decrease promoter activity. The TFIIB DNA consensus binding sequence is 5’-G/C-G/C-G/A-C-G-C-C-3’, and the upstream TFIIB site has 6 of 7 residues conserved, while the downstream site has only 3 of 7 residues conserved. This suggests that the upstream site is more likely to be the true TFIIB binding site, which explains why deletion of the upstream TFIIB site results in decreased promoter activity.
It is interesting that deletion of both predicted TFIIB sites does not completely abolish promoter activity, but only reduced activity by approximately 32% (Figure 7). This suggests that other TFIIB sites are present within the -1386 bp sequence that allows recruitment and assembly of the RNA pol II complex. Recent studies show that DNA binding by TFIIB is more complicated than originally proposed, because TFIIB has two independent DNA-recognition motifs that recognize different DNA consensus sequences [25,26]. The previously well-characterized DNA-recognition motif within TFIIB binds to DNA through a Helix-Turn-Helix motif (HTH), and its preferred binding consensus sequence is partially conserved in 34% of eukaryotic promoters, which is located upstream of the TATA box [27]. However the second DNA-recognition motif of TFIIB binds to DNA through a loop region between two alpha-helices of TFIIB, and unlike the HTH motif, its DNA consensus sequence is located downstream of the TATA box [25,26]. This TFIIB DNA-recognition motif is likely as important as the HTH motif, since its preferred DNA consensus sequence is partially conserved in up to 37% of eukaryotic promoters [25,26]. It has a loosely defined DNA consensus sequence, 5’-G/A-T-T/G/A-T/G-G/TT/G-T/G-3’, that is less specific than the consensus sequence of the HTH motif [25,26]. Therefore, although we deleted both predicted TFIIB binding sites upstream of the TATA box in the RDH10 promoter, the residual promoter activity may be explained by the presence of a novel unpredicted TFIIB binding site downstream of the TATA box in the RDH10 promoter that is recognized by the non-HTH DNA-recognition motif within TFIIB. Our sequence analyses did not find such a DNA consensus sequence in the RDH10 proximal promoter; however, previous studies have shown that as little as 3 of the 7 base pairs of the DNA consensus sequence can be sufficient for binding to TFIIB [25], so it is possible that there is another cryptic TFIIB binding site present in the RDH10 promoter.
The protein-DNA interaction observed using the 167-bp probe in EMSA studies may represent a large multi-protein complex that includes other transcription factor(s) in addition to TFIIB, including other components of the RNA pol II complex. It is also possible that other zinc-dependent and independent transcription factors are present, although zinc-dependent transcription factors appear to be most crucial for stabilizing the protein-DNA interaction since zinc must be available in order to observe the complex by EMSA. Other zinc-dependent transcription factor-binding sites predicted within the 167-bp sequence include several krueppel like factors, such as zinc finger protein MOK2 (MOK2), basic and erythroid Krueppel Like Factors (KLF3 and KLF15), and gut-enriched Krueppel Like Binding Factor (KLF4). Interestingly, KLF15 has been shown previously to act as a transcriptional repressor for two retina-specific genes, rhodopsin and Interphotoreceptor Binding Protein (IRBP) [28], and MOK2 has also been shown to mediate transcriptional repression of IRBP [29]. Predicted zinc-independent transcription factor binding sites include NKX homeodomain factors (NKX25 and NKX32), which are known to be important for regulating tissue-specific gene expression to coordinate organ development during embryogenesis [30].
In summary, the data presented herein demonstrated for the first time the basal proximal promoter sequence of the human RDH10 gene. This analysis identifies a 92-bp sequence between -756 bp and -848 bp upstream of the TSS that is essential for basal promoter activity. This 92-bp sequence contains a TFIIB-binding site that is necessary for maximum promoter activity. Further studies are necessary to determine if there is a second novel TFIIB site downstream of the TATA box, and to identify tissue-specific transcription factors that may serve to regulate RDH10 expression during embryogenesis and in adult RPE and retinal Muller cells.
Acknowledgement
The research reported in this study was supported by the Oklahoma Center for the Advancement of Science and Technology (Grant #HR13-075; Principal Investigator K.M.F.).
References
- R, Blomhoff HK. Overview of retinoid metabolism and function. J Neurobiol. 2006; 66: 606-630.
- Clagett-Dame M, DeLuca HF. The role of vitamin A in mammalian reproduction and embryonic development. Annu Rev Nutr. 2002; 22: 347-381.
- Thompson DA, Gal A. Vitamin A metabolism in the retinal pigment epithelium: genes, mutations, and diseases. Prog Retin Eye Res. 2003; 22: 683-703.
- Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007; 47: 469-512.
- Sandell LL, Sanderson BW, Moiseyev G, Johnson T, Mushegian A, Young K, et al. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007; 21: 1113-1124.
- Farjo KM, Moiseyev G, Nikolaeva O, Sandell LL, Trainor PA, Ma JX. RDH10 is the primary enzyme responsible for the first step of embryonic Vitamin A metabolism and retinoic acid synthesis. Dev Biol. 2011; 357: 347-355.
- Cammas L, Romand R, Fraulob V, Mura C, Dolle P. Expression of the murine Retinol dehydrogenase 10 (Rdh10) gene correlates with many sites of retinoid signaling during embryogenesis and organ differentiation. Dev Dyn. 2007; 236: 2899-2908.
- Romand R, Kondo T, Cammas L, Hashino E, Dolle P. Dynamic expression of the retinoic acid-synthesizing enzyme retinol dehydrogenase 10 (rdh10) in the developing mouse brain and sensory organs. J Comp Neurol. 2008; 508: 879-892.
- Wu BX, Chen Y, Chen Y, Fan J, Rohrer B, Crouch RK, et al. Cloning and characterization of a novel all-trans retinol short-chain dehydrogenase/reductase from the RPE. Invest Ophthalmol Vis Sci. 2002; 43: 3365-3372.
- Wu BX, Moiseyev G, Chen Y, Rohrer B, Crouch RK, Ma JX. Identification of RDH10, an All-trans Retinol Dehydrogenase, in Retinal Muller Cells. Invest Ophthalmol Vis Sci. 2004; 45: 3857-3862.
- Kiser PD, Golczak M, Maeda A, Palczewski K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim Biophys Acta. 2012; 1821: 137-151.
- Saari JC. Vitamin A metabolism in rod and cone visual cycles. Annu Rev Nutr. 2012; 32: 125-145.
- Tang PH, Kono M, Koutalos Y, Ablonczy Z, Crouch RK. New insights into retinoid metabolism and cycling within the retina. Prog Retin Eye Res. 2013; 32: 48-63.
- Farjo KM, Moiseyev G, Takahashi Y, Crouch RK, Ma JX. The 11-cis-retinol dehydrogenase activity of RDH10 and its interaction with visual cycle proteins. Invest Ophthalmol Vis Sci. 2009; 50: 5089-5097.
- Picozzi P, Marozzi A, Fornasari D, Benfante R, Barisani D, Meneveri R, et al. Genomic organization and transcription of the human retinol dehydrogenase 10 (RDH10) gene. FEBS Lett. 2003; 554: 59-66.
- Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006; 41: 105-178.
- Reese JC. Basal transcription factors. Curr Opin Genet Dev. 2003; 13: 114-118.
- Kostrewa D, Zeller ME, Armache KJ, Seizl M, Leike K, Thomm M, et al. RNA polymerase II-TFIIB structure and mechanism of transcription initiation. Nature. 2009; 462: 323-330.
- Blanco JC, Wang IM, Tsai SY, Tsai MJ, O'Malley BW, Jurutka PW, et al. Transcription factor TFIIB and the vitamin D receptor cooperatively activate ligand-dependent transcription. Proc Natl AcadSci USA. 1995; 92: 1535-1539.
- Leong GM, Wang KS, Marton MJ, Blanco JC, Wang IM, Rolfes RJ, et al. Interaction between the retinoid X receptor and transcription factor IIB is ligand-dependent in vivo. J Biol Chem. 1998; 273: 2296-2305.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72: 248-254.
- Elsby LM, Roberts SG. Interaction of the TFIIB zinc ribbon with RNA polymerase II. Biochem Soc Trans. 2008; 36: 595-598.
- Ghosh M, Elsby LM, Mal TK, Gooding JM, Roberts SG, Ikura M. Probing Zn2+-binding effects on the zinc-ribbon domain of human general transcription factor TFIIB. Biochem J. 2004; 378: 317-324.
- Chen HT, Hahn S. Binding of TFIIB to RNA polymerase II: Mapping the binding site for the TFIIB zinc ribbon domain within the preinitiation complex. Mol Cell. 2003; 12: 437-447.
- Deng W, Roberts SG. A core promoter element downstream of the TATA box that is recognized by TFIIB. Genes Dev. 2005; 19: 2418-2423.
- Deng W, Roberts SG. Core promoter elements recognized by transcription factor IIB. Biochem Soc Trans. 2006; 34: 1051-1053.
- Lagrange T, Kapanidis AN, Tang H, Reinberg D, Ebright RH. New core promoter element in RNA polymerase II-dependent transcription: sequence-specific DNA binding by transcription factor IIB. Genes Dev. 1998; 12: 34-44.
- Otteson DC, Liu Y, Lai H, Wang C, Gray S, Jain MK, et al. Kruppel-like factor 15, a zinc-finger transcriptional regulator, represses the rhodopsin and interphotoreceptor retinoid-binding protein promoters. Invest Ophthalmol Vis Sci. 2004; 45: 2522-2530.
- Arranz V, Dreuillet C, Crisanti P, Tillit J, Kress M, Ernoult-Lange M. The zinc finger transcription factor, MOK2, negatively modulates expression of the interphotoreceptor retinoid-binding protein gene, IRBP. J Biol Chem. 2001; 276: 11963-11969.
- Stanfel MN, Moses KA, Schwartz RJ, Zimmer WE. Regulation of organ development by the NKX-homeodomain factors: an NKX code. Cell Mol Biol (Noisy-le-grand). 2005; 51: OL785-799.