
Case Report
J Ophthalmol & Vis Sci. 2021; 6(1): 1045.
Ciliary Ganglioplegic Migraine Associated with SACS Mutation
Kang JJ1, Lee HM1, Ki CS3 and Oh SY1,2*
¹Department of Neurology, Jeonbuk National University, Republic of Korea
²Jeonbuk National University, Research Institute of Clinical Medicine, Republic of Korea
³Green Cross Genome, Republic of Korea
*Corresponding author: Sun-Young Oh, Department of Neurology, National University, Research Institute of Clinical Medicine, Jeonbuk 20 Geonji-ro, Deokjin-gu, Jeonju-city, Jeonbuk, 561-712, South Korea
Received: December 02, 2020; Accepted: February 04, 2021; Published: February 11, 2021
Case Presentation
Migraine patients often complain of repeated attacks of monocular visual disturbance including scintillations, scotomas, blindness and pupillary dysfunction. Temporary pupillary enlargement of the ipsilateral side during migraine attack, although rarely reported, is referred to as an Adie’s-like tonic pupil [1]. Adie’s tonic pupil is caused by denervation of the postganglionic parasympathetic pupillary nerves, with light-near dissociation, and cholinergic supersensitivity.
Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) is characterized by early-onset spastic ataxia, peripheral sensorimotor neuropathy, and pyramidal symptoms. ARSACS is caused by mutations in the SACS gene, which encodes the Sacsin expressed in various tissues including brain motor systems. Sacsin is believed to integrate the ubiquitin-proteasome system and Hsp70 chaperone machinery, which are important components of the cellular response associated with neurodegenerative diseases [2].
Here, we describe a patient suffering from chronic migraine without aura who developed cerebellar ataxia and unilateral mydriasis during acute migraine persisted after resolved headache. Presence of a SACS gene mutation is an expansion of the clinical phenotype associated with SACS mutations to ciliary ganglioplegic migraine.
A 47-year-old female who suffers from long standing headache developed unsteady gait and poor coordination. With a diagnosis of migraine without aura based on the third edition of the International Classification of Headache Disorders (ICHD-III) criteria, she was prescribed preventive medicine with topiramate and onabotulinumtoxinA injection on occasion. Physical examination revealed a dilated pupil in the left eye without reflexive constriction to light or accommodation which persisted after resolution of migraine. Except for anisocoria, no other abnormal findings were identified. Remarkable reduction of the left pupil size was observed in response to 0.125% pilocarpine (Figure 1). The patient also showed mild nonataxic spastic paraplegia but without distal weakness or pyramidal signs. A family history of migraine headache and other neurological disorders was denied. Nerve conduction studies confirmed the mixed demyelinating and axonal character of polyneuropathy. Brain MRI with angiography did not reveal any abnormalities.
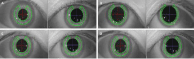
Figure 1: Pupillary response in the light (A; 7.2 mm and 8.7 mm in the right and left pupils, respectively), in darkness (B; 8.6 mm and 9.6 mm) and during
accommodation (D; 7.2 mm and 8.4 mm). Supersensitivity to 0.125% pilocarpine was observed in the light (C; 7.2 mm and 7.4 mm).
As other ataxia and migraine disorders could not be excluded, genes potentially associated with the current symptoms were carefully examined. Genetic tests for spinocerebellar ataxias 1, 2, 3, 6, and 7 were negative. Genomic DNA was extracted from peripheral blood leukocytes. Targeted ataxia gene panel testing was performed at GC Genome (Yongin, Gyeonggi-do, Korea). A customized target enrichment system kit (Celemics, Seoul, Korea) was used to capture each exon for 30 ataxia-related genes (ADCK3, ANO10, APTX, ATM, C10orf2, CACNA1A, CACNB4, CYP27A1, FGF14, ITPR1, KCNA1, KCNC3, KCND3, PEX7, PNKP, PNPLA6, POLG, PRKCG, SACS, SETX, SIL1, SLC52A2, SNX14, SPG7, SPTBN2, SYNE1, TMEM240, TTBK2, TTPA, WFS1) and 147 additional genes (available upon request) spanning a 616,206 bp region. Sequencing was performed using the MiSeqDx platform (Illumina Inc., San Diego, CA, USA) with 150-bp paired-end sequencing. Alignment of sequence reads, indexing of the reference genome (hg19), and variant calling with a pipeline were performed based on GATK best practices. A novel compound heterozygous mutation in the SACS gene was identified in our patient (c.6860A>G and p.Lys2287Arg) (Figure 2).

Figure 2: A heterozygous c.6860A>G variant in the SACS gene (arrow) viewed on Genome Browse 3.0.0 (Golden Helix Inc., Bozeman, MT). About 50% of the
parts that should normally consist of T sequence have been changed to G (yellow) sequence.
The patient was treated with onabotulinumtoxinA injection and preventive medications of topiramate with good clinical response. Despite cessation of migraine attacks, dilated pupil on the left side persisted. For tonic pupil, 0.125% pilocarpine was prescribed, and the patient was discharged.
In the current study, we report a patient with ciliary ganglioplegic migraine and a concurrent, novel compound heterozygous mutation in the SACS gene. Several studies have reported ipsilateral pupillary dilatation during acute migraine attacks; however, mydriasis outlasting the headache is very rare and has been described as a comorbidity of migraine and Adie’s tonic pupil caused by denervation of the post-ganglion supply to the pupillary sphincter and ciliary muscle. However, the association between Adie’s tonic pupil and migraine, the ‘ciliary ganglioplegic migraine,’ has not been clearly established. As the ciliary ganglia are distributed in the nasal branch of the ophthalmic division (V1) of the trigeminal nerve, the ciliary ganglia is stimulated during migraine attack based on the neurovascular theory of migraine. Therefore, the migraine leads to ipsilateral ciliary ganglion cell dysfunction and results in pupil dilatation. A previous report suggested a link between migraine and autonomic ciliary ganglionopathy and peripheral neuropathy [3], where autonomic ganglionitis and peripheral nerve inflammation were confirmed through autopsy of patients with recurrent migraine-induced hypotension and visual impairment. Another theory is that ophthalmoplegic migraine with localized lesions of the parasympathetic fibers of the ocular motor nerve can cause migraine headache and ipsilateral mydriasis. However, the degree of cholinergic hypersensitivity of the iris sphincter during the pilocarpine test cannot distinguish preganglionic mydriasis from ganglionic or postganglionic Adie’s pupil. Although, there is no clear explanation for the persistent mydriasis, it may be presumed that repetitive attacks cause damage to the pupillary nerve fibers, resulting in persistent mydriasis even after headache resolution.
Recently, SACS mutations were found in atypical ARSACS such as delayed onset ataxia, non-ataxic spastic paraplegia, mild pyramidal signs, and supratentorial abnormalities [4]. Sacsin, encoded by the SACS gene, is located on the surface of the mitochondria and is involved in its metabolism. SACS mutations in ARSACS patients decrease the rate of the mitochondrial respiratory chain linked to ATP production, thereby inhibiting metabolic activity and energy production and causing bioenergetic damage to cells [5]. As a result, it may promote or contribute to apoptosis occurring in the neurodegenerative processes of nervous system by inducing oxidative stress and defects in bioenergetic processes. Due to damage to the central and peripheral nervous systems through this series of processes, symptoms such as cerebellar atrophy and ataxia caused by the loss of Purkinje fibers in the cerebellum, spinal cord atrophy, and peripheral neuropathy, which are representative clinical symptoms of ARSACS, are produced. Migraine with pupillary dysfunction has never been documented in ARSACS. Herein, we describe a patient bearing a novel SACS mutation who presented with ARSACS and ciliary ganglioplegic migraine. Patients with SACS mutations may manifest a ciliary ganglioplegic migraine phenotype with typical symptoms of ARSACS. These findings indicate SACS is likely the causal gene of the combined syndrome and the ARSACS phenotype is variable and should be biologically validated.
Acknowledgement
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of Science and ICT) (No. 2019R1A2C1004796).
References
- Skeik N, Jabr FI. Migraine with benign episodic unilateral mydriasis. Int J Gen Med. 2011; 4: 501-503.
- Parfitt DA, Prodromou NV, Webb TR, Gallo J-M, Cheetham ME, Nicoll WS, et al. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Human Molecular Genetics. 2009: 18: 1556-1565.
- Lee H-C, Coulter CL, Adickes ED, Porterfield J, Robertson D, Bravo E, et al. Autonomic ganglionitis with severe hypertension, migraine, and episodic but fatal hypotension. Neurology. 1996: 47: 817-821.
- Baets J, Deconinck T, Smets K, Goossens D, den Bergh PV, Schmedding E, et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology. 2010; 75: 1181-1188.
- Criscuolo C, Procaccini C, Meschini MC, Cianflone A, Carbone R, Doccini S, et al. Powerhouse failure and oxidative damage in autosomal recessive spastic ataxia of Charlevoix-Saguenay. J Neurol. 2015; 262: 2755-2763.