
Research Article
J Ophthalmol & Vis Sci. 2021; 6(2): 1049.
Visual Acuity and Age of Symptom Onset in a Large Chinese Cohort of Patients with Stargardt Disease
Gao F-J1,2,3#, Wang D-D1,2,3#, Hu F-Y1,2,3, Xu P1,2,3, Chang Q1,2,3, Liu W1,2,3, Zhang S-H1,2,3, Huang Y1, Xu G-Z1,2,3* and Wu J-H1,2,3*
1Eye Institute, Eye and ENT Hospital, College of Medicine, Fudan University, Shanghai, China
2Shanghai Key Laboratory of Visual Impairment and Restoration, Science and Technology Commission of Shanghai Municipality, Shanghai, China
3Key Laboratory of Myopia (Fudan University), Chinese Academy of Medical Sciences, National Health Commission, China
#Contributed Equally to this Work
*Corresponding author: Ji-Hong Wu, Eye and ENT Hospital, Fudan University, Shanghai, 83 Fengyang Road, Shanghai, 200031, China
Ge-Zhi Xu, Eye and ENT Hospital, Fudan University, Shanghai, 83 Fengyang Road, Shanghai, 200031, China
Received: April 22, 2021; Accepted: May 20, 2021; Published: May 27, 2021
Abstract
Stargardt disease is the most common form of juvenile-onset inherited macular dystrophy, with high phenotypic heterogeneity. There are limited data documenting the characteristics of Best Corrected Visual Acuity (BCVA) and age of symptom onset of patients with Stargardt disease in Chinese population. In this study, 169 patients with clinical and genetic diagnosis of Stargardt disease were recruited. BCVA, age, age of symptom onset, disease duration time and genetic information were collected and analyzed. We found that the average BCVA was 1.16±0.61 logMAR, 63.6% of patients with severe visual impairment (≤1 logMAR); the mean age of symptom onset was 13.63±12.72 years (yrs) old, 61.36% of patients had an onset earlier than 12; BCVA level was associated with genotype and disease duration, but had no direct correlation with neither age of symptom onset nor age; severe genotype (≥2 severe variants, severe variants were defined as those predicted to introduce a premature truncating codon in the protein or to affect splicing) was also associated with earlier age of symptom onset. Our data will serve as a well-founded reference for genetic counseling and better management of these patients.
Keywords: Stargardt disease; ABCA4 gene; Chinese population; BCVA; Genotype; Age at symptom onset; Disease duration; Age
Abbreviations
BCVA: Best Corrected Visual Acuity Testing; SD-OCT: Spectral Domain Optical Coherence Tomography
Introduction
Stargardt disease is an autosomal recessive retinal dystrophy mainly caused by mutations in the ATP Binding Cassette Subfamily A Member 4 Gene (ABCA4), and a small proportion is caused by PROM1, PRPH2, ELOVL4 mutations [1-3]. It is the most common form of inherited macular dystrophy onset from birth and is characterized by bilateral degeneration of the central retina, and a beaten-bronze aspect of the retina seen during funduscopy with or without yellow-white flecks in the posterior pole [4,5]. The reported prevalence is 1 in 8000-10,000 individuals [6,7].
Stargardt disease has a highly heterogeneous phenotype, ranging from mild to extremely aggressive forms [7-10]. Some patients have disease onset from birth, while others develop disease after the age of 50 years [11,12]. Additionally, although some patients show a rapid loss of Visual Acuity (VA) of 0.066 logMAR per year, some maintain a relatively good VA for decades [13,14]. The VA can also vary in patients with Stargardt disease, ranging from 20/20 to 20/400 or worse [4,15].
Several studies have reported changes in VA loss in Stargardt disease and associated risk factors [13-20]. One of the most famous of these is the ProgStar study, which reported that VA change is associated with foveal phenotype and declines faster in younger patients, but is not associated with genotype [13,18]. However, descriptions of the clinical characteristics of Stargardt disease varied between studies, which may reflect differences in study methods. For example, ProgStar study report No. 10 showed that genotype was neither associated with baseline Best Corrected Visual Acuity (BCVA) nor with the rate of BCVA change [13], while Traboulsi et al reported that patients who harboured two or more mutations presented with disease earlier and had a worse BCVA than those with no or one mutation [20].
Limited data are available about the characteristics of VA and age of symptom onset in Chinese patients with Stargardt disease. Therefore, this study aimed to determine the basic characteristics of Chinese patients with Stargardt disease, including VA, age of symptom onset, and genotype, and to evaluate possible correlations among them.
Methods
Subjects and ethical statement
This is a retrospective observational study approved by the Ethics Committee of the Eye and ENT Hospital of Fudan University, which adheres to the Declaration of Helsinki. A total of 169 patients with a definitive clinical and genetic diagnosis of Stargardt disease were recruited from our ophthalmology department between January 2015 and November 2019. A clinical diagnosis was based on one or more of the following features as reported previously [14,26,27]; macular changes such as atrophy or a beaten-bronze metal appearance, and the presence of a well-defined atrophic lesion with or without characteristic white-yellow flecks at the level of the retinal pigment epithelium involving the posterior pole or extending to the midperipheral retina flecks on fundus auto fluorescence studies. The main genetic diagnostic criteria were at least two likely disease causing ABCA4 variants or one or more likely disease-causing PROM1 variant associated with a typical Stargardt disease phenotype. Exclusion criteria included any other ocular diseases that may confound morphologically and functional assessment of the retina, ophthalmologic surgery within the previous 90 days, any systemic condition potentially affecting the analyses, and previous or current participation in any drug trial or clinical trial to treat Stargardt disease. Written informed consent was obtained from all participants and/or legal guardians before peripheral blood samples were collected.
Genetic analysis
Molecular testing and data analysis was performed as previously reported [28-32]. We designed a high-through put targeted enrichment approach to exon-capture regions of 762 genes involved in common inherited eye diseases (BGI, Shenzhen, China). For acquisition of probe sequences, the exon sequence and its flank ±30bp from a reference human genome (UCSC hg 38) was obtained. Each reference sequence begins at one end of a reference sequence and selects the reference sequence of the predetermined length to obtain the probe sequence so that the last total probe can cover the reference sequence at least once. The probe length of the panel is 90nt, the total target area obtained is 2.3M. On average, the mean coverage depth was more than 400X, and the coverage of target region was ~99.9% using BGISEQ-2000. Subsequent points for sample quality control were also added to the probe design process. The following databases were then used for annotation of all identified variants with Minor Allele Frequency (MAF) >0.1% to eliminate benign variants: dbSNP137, HapMap Project, 1000 Genomes Project, YH database4, ESP6500, and Exome Variant Server. Finally, the variant prioritizations were performed, combining total depth, quality score, MAF, potential deleterious effect (five prediction algorithms: SIFT, Polyphen2, LRT, Mutation Taster, and FATHMM) and the existence of mutation reports in common databases such as the Human Gene Mutation Database (HGMD, professional version 2020 [4]), ClinVar, Retinal Information Network (RetNet), or Online Mendelian Inheritance in Man (OMIM). The prediction of novel missense variants is listed in Supplementary Table 2. Variants were classified as pathogenic, likely pathogenic, and novel variants of uncertain clinical significance according to the American College of Medical Genetics. Based on the pathogenicity, number, and severity of the ABCA4 or PROM1 mutations identified in each patient, patients harbouring two or more mutations were divided into three groups: genotype A group (severe): patients with ≥2 severe variants; genotype B group: patients with one severe variant and one missense mutation or in-frame insertion/deletion; and the genotype C group (moderate): patients with no severe variant but ≥2 missense variants or in-frame insertions/deletions. Severe variants were defined as those predicted to introduce a premature truncating codon in the protein or to affect splicing, such as frameshift, nonsense, or intronic or exonic variants causing major splice site alterations [11,13].
Clinical examination
All participants underwent a full ophthalmologic examination including the following assessments: best Snellen-corrected visual acuity testing (converted to an equivalent value of the logarithm of the minimal angle of resolution (logMAR) unit), slitlamp biomicroscopy of anterior and posterior segments, sweptdomain optical coherence tomography (Spectralis HRA+OCT, Heidelberg Engineering Inc., Heidelberg, Germany), and full-field electroretinography according to the standards of the International Society for Clinical Electrophysiology of Vision (www.iscev.org). The following conversions were made to provide numeric values for low BCVAs: hand motion (HM), 3 logMAR; and counting fingers, 2 logMAR [33,34]. Age at symptom onset was defined as the age when the patient first experienced symptoms associated with the disease (VA decline, loss of colour vision, and central and paracentral visual field defects), while disease duration was calculated by subtracting the onset date from the examination date. Right eye was used for VA analysis.
Statistical analysis
Measurement values of the groups were compared using the t-test and one-way analysis of variance test. Correlations were evaluated using Pearson, Spearman, or linear regression analyses. Statistical analyses were performed using SPSS version 20.0 software (SPSS Inc./IBM Corp., Chicago, IL) and Microsoft Excel (2010). Statistical significance for all tests was set at p <0.05.
Results
Cohort characteristics
A total of 169 patients of Chinese origin with clinically and molecularly confirmed Stargardt disease were enrolled (Supplementary Table 1). Table 1 summarizes baseline demographic and clinical characteristics of all participants. The 169 patients were from 157 families and included 102 men (60.4%) and 67 women (39.6%) with an age range of 5-64 years (mean, 28.28 ± 14.56 years; median, 26 years).
![]()
Characteristics
Data
No. patients (%)
Total
169 (100%)
Female/male
67/102 (39.6%/60.4%)
Age =10 yrs
18 (10.7%)
Age 10-20 yrs
43 (25.4%)
Age 20-50 yrs
98 (58.0%)
Age >50yrs
10 (5.9%)
Genotype A
28 (17.4%)
Genotype B
68 (42.2%)
Genotype C
65 (40.4%)
*Onset =5 yrs
36 (27.3%)
Onset 6-12 yrs
45 (34.1%)
Onset 13-20 yrs
17 (12.9%)
Onset 21-49 yrs
33 (25.0%)
Onset =50 yrs
1 (0.7%)
No. eyes (%)
338
#HM-1.3 (BCVA)
134 (39.6%)
1.3-1 (BCVA)
81 (24.0%)
1-0.52 (BCVA)
94 (27.8%)
0.52-0.3 (BCVA)
18 (5.3%)
0.3-0 (BCVA)
11 (3.3%)
BCVA logMAR
Average
1.16±0.61
Age =10yrs
0.77±0.25
Age 10-20 yrs
0.99±0.37
Age 20-50 yrs
1.29±0.68
Age >50yrs
1.33±0.77
Disease duration =10yrs
0.90±0.40
Disease duration 10 -20 yrs
1.12±0.37
Disease duration 10 -20 yrs
1.46±0.53
Disease duration >20yrs
1.73±0.84
Genotype A
1.38±0.75
Genotype B
1.20±0.51
Genotype C
1.07±0.65
Age of onset (yrs)
Average
13.63±12.72
Genotype A
5.85±6.82
Genotype B
13.82±11.44
Genotype C
14.92±13.11
Mean disease duration (yrs)
13.74±11.85
No: Number; BCVA: Best-Corrected Visual Acuity; HM: Hand Moving; yrs: Years. *Age of onset was available for 132 patients; #BCVA was available in all patients (N=169).
Table 1: Baseline demographic and clinical characteristics of all participants.
BCVA
BCVA was available for all patients and was shown to be similar between both eyes (p=0.753, t=0.315, df=338), with means of 1.15 ± 0.60 logMAR and 1.16 ± 0.63 logMAR for right and left eyes, respectively. Most patients had a binocular BCVA difference of less than 0.70 logMAR (40/200); only 20 (11.83%, 20/169) had a binocular BCVA difference ≥0.70 logMAR. Measured BCVA ranged from 0 logMAR (20/20) to HM; 39.64% (134/338) of eyes had a BCVA worse than logMAR 1.3, 63.61% (215/338) had a BCVA worse than 1 logMAR, 91.42% (309/338) had a BCVA worse than 0.52 logMAR, and only 29 eyes (8.58%) had a BCVA better than 0.52 logMAR (Figure 1A and Table 1).
BCVA correlations with age
LogMAR BCVA versus age of all patients is depicted in Figure 1B, with the Snellen equivalent shown on the right. We observed a positive correlation of logMAR BCVA with age (Pearson’s coefficient: 0.241, p <0.001), in which older age was associated with a significantly worse BCVA. Patients were then divided into four age groups: ≤10 years old (n=36, mean BCVA 0.77 ± 0.25 logMAR), 10-20 years (n=86, median BCVA 0.99 ± 0.37 logMAR), 20-50 years (n=196, median BCVA 1.29 ± 0.68 logMAR), and >50 years (n=20, median BCVA 1.33 ± 0.77 logMAR). BCVA data according to age group are shown in Table 1. The BCVA in patients younger than 10 years was significantly better than in those aged 10-20 years (p<0.001), 20-50 years (p<0.001), and >50 years (p<0.05), while the BCVA in patients aged 10-20 years was significantly better than in patients aged 20-50 years (p<0.001), but not with patients >50 years (p>0.05). There were also no significant differences in BCVA between patients aged 20-50 years and >50 years (p>0.05; Figure 1B). BCVA distributions in patients of the four age groups are shown in Figure 1C and Table 1. None of the patients ≤10 years of age had a BCVA worse than 1.3 (logMAR BCVA), while the proportion of patients with legal blindness (HM–1.3 logMAR BCVA) increased with age. The data were normalized to eliminate the confounding effect of disease durations and genotype, which ensured that all patients had a common baseline to conduct a meaningful t test analysis of BCVA and age. After adjusting, age was no longer significantly associated with BCVA changes (p=0.192).
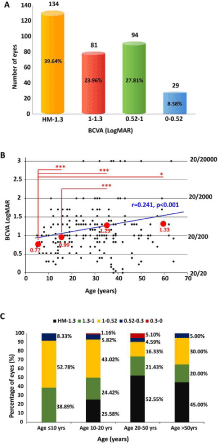
Figure 1: A) Number of eyes in different best-corrected visual acuity
(BCVA, logMAR) groups. HM-1.3 logMAR group: 134 eyes (39.64%); 1-1.3
logMAR group: 81 eyes (23.96%); 0.52-1 logMAR group: 94 eyes (27.81%);
0-0.52 logMAR group: 29 eyes (8.58%). B) Correlation of age with bestcorrected
visual acuity (BCVA, logMAR) (coefficient: 0.241, p<0.001).
Snellen equivalent is displayed on the right. The red circles represent BCVA
in different age groups: age ≤10 years old (yrs), median BCVA 0.77±0.25
logMAR; age 10-20 yrs, median BCVA 0.99±0.37 logMAR; age 20-50 yrs,
median BCVA 1.29±0.68 logMAR; age >50yrs, median BCVA 1.33±0.77
logMAR; ***p<0.001; *p<0.05. C) Distributions of best-corrected visual acuity
(BCVA, logMAR) in patients of different age groups.
BCVA correlations with age at symptom onset and disease duration
Age at symptom onset was available for 132 patients. The mean age at symptom onset was 13.63 ± 12.72 years (range, 0-54 years; median, 9 years), and the mean disease duration was 13.74 ± 11.85 years (range, 1-63 years; median, 10 years). We found that 27.27% (n=36) of patients had disease onset before 5 years of age (infant onset), 34.09% (n=45) were between 6-12 years (juvenile onset), 12.88% (n=17) were between 13-20 years, 25% (n=33) were between 21-49 years, and only one (0.76%) had disease onset >50 years of age (Figure 2A). Both the age at symptom onset (Pearson’s coefficient -0.271, Figure 2B) and disease duration (Pearson’s coefficient 0.589, Figure 2C) had a significant association with BCVA (p<0.001) when analyzed separately. Data were normalized because the age at onset and disease duration are not independent of each other. The normalised disease duration was significantly correlated with BCVA, with a longer disease duration being associated with a worse BCVA (coefficient: 0.559, p<0.001). However, no association was evident between BCVA and age at symptom onset (p=0.212).
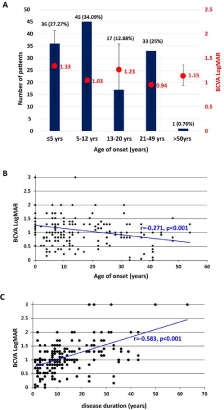
Figure 2: A) Number of patients in different age of symptom onset groups.
≤5 yrs: 36 patients (27.27% ); 6-12 yrs: 45 patients (34.09%); 13-20 yrs: 17
patients (12.88%); 21-49 yrs: 33 patients (25%); ≥50yrs: 1 patients (0.76%).
Best-corrected visual acuity (BCVA, logMAR) is displayed on the right in
red font. The red circles represent mean BCVA in different age of symptom
onset groups. B) Correlations between best-corrected visual acuity (BCVA,
logMAR) values and ages of symptom onset (coefficient: -0.271, p<0.001).
C) Correlations between best-corrected visual acuity (BCVA, logMAR) values
and disease duration (coefficient: -0.583, p<0.001).
BCVA correlations with genotypes
ABCA4 mutations were identified in 150 patients and PROM1 mutations were identified in 19. A total of 171 unique variants were identified of which 64 were novel. Of the 169 patients, 161 had compound heterozygous or homozygous mutations, and eight displayed dominant inheritance with only one PROM1 allele identified. The 161 patients who harboured two or more variants were divided into genotype A (n=28), genotype B (n=68), and genotype C groups (n=65), with mean values of 1.38 ± 0.75, 1.20 ± 0.51, and 1.07 ± 0.65 logMAR BCVA, respectively. There was no significant difference between these values (p>0.05; Figure 3A), but correlation analysis showed that genotype A was associated with a significantly worse BCVA (Spearman’s coefficient: -0.184, p<0.01, Figure 3B).
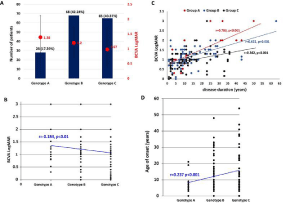
Figure 3: A) Number of patients in different genotype groups. Genotype A group: 28 patients (17.39%), genotype B group: 68 patients (42.24%), genotype C group:
65 patients (40.37%). Best-corrected visual acuity (BCVA, logMAR) is displayed on the right in red font. The red circles represent mean BCVA in different genotype
groups.
B. Correlations between best-corrected visual acuity (BCVA, logMAR) values and genotype (coefficient: 0.184, p<0.01). C) Scatterplot of logMAR BCVA versus
disease duration in the three genotype groups. BCVA decreased with disease duration prolongs in all the three groups, but BCVA decrease faster in group A (red
dots, Pearson’s coefficient: 0.783, P <0.001), relatively slow in group B (blue dots, Pearson’s coefficient: 0.622, P <0.001), and slowest in group C (black dots,
Pearson’s coefficient: 0.362, P <0.001). D) Correlations between best-corrected visual acuity (BCVA, logMAR) values and genotype (coefficient: 0.237, p <0.001).
When disease duration data were normalized, a significant association was still observed (coefficient: -0.274, p<0.001) which was confirmed by a scatterplot of logMAR BCVA versus disease duration in the three genotype groups (Figure 3C). BCVA decreased with the length of disease duration in all three genotype groups, but the decrease was most rapid in group A (Pearson’s coefficient: 0.783, p <0.001), relatively slow in group B (Pearson’s coefficient: 0.622, p <0.001), and slowest in group C (Pearson’s coefficient: 0.362, p <0.001). The mean age at symptom onset in the three groups was 5.85 ± 6.82, 13.82 ± 11.44, and 14.92 ± 13.11 years, respectively. Patients in group A had a significantly earlier symptom onset than those in groups B and C (p<0.001), indicating that this may be associated with genotype. Spearman analysis revealed a positive correlation between age at symptom onset and genotype (coefficient: 0.237, p <0.001), with genotype A being associated with a significantly earlier onset (Figure 3D).
Discussion
Before data normalization, patient age, age at symptom onset, and disease duration were all significantly associated with BCVA. However, after data normalization, the association between BCVA and age disappeared, which could be explained by the fact that older patients often have a longer duration of disease. ProgStar Study Reports 6 and 2 found that older age at symptom onset was associated with a better BCVA, and that an earlier symptom onset and longer duration of disease were associated with a worse BCVA [17,18]. These findings do not contradict with our own because we showed that BCVA was significantly associated with genotype, which itself was associated with age at symptom onset. It is likely that the influence of age at symptom onset on BCVA is secondary to the genotype. Specific genotype information was not recorded in the ProgStar study, so further genotype analyses were not performed. We not only identified a correlation between BCVA and age at symptom onset, but also clarified the phenomenon found in ProgStar Study Reports 6 and 2 from the perspective of pathogenesis. Furthermore, our results are also in agreement with those of a retrospective, cross-sectional study of 112 patients with Stargardt disease from 2014 which showed that BCVA is not associated with age at disease presentation [20].
Understanding the genotype-phenotype correlation is challenging because of the large number of ABCA4 mutations. Moreover, because most patients are compound heterozygotes, it is even more difficult to assess the effect of each mutation on phenotype and to evaluate possible allelic hierarchy. Multiple studies have investigated genotype– phenotype correlations in Stargardt disease [4,20-23] but the results are controversial and 86% of the participants of this study were white and likely had different genetic backgrounds to Chinese patients. ProgStar Study Report No. 10 showed that baseline BCVA and BCVA change rates were not associated with genotype [13], but study participants were enrolled from multiple sites in the United States and Europe, and 86% were white while all of our participants were of Chinese origin. Differences in ethnicities may lead to varied results. For example, the prevalence of genotype A was only 3% (n=8) in the ProgStar cohort, compared with 17.4% in our study. Additionally, patients with only one variant predicted as less likely to be pathogenic or uncertain (genotype D) were also included for analysis in the ProgStar study, which may have affected the results. Because patients with genotype D did not have a definitive diagnosis of Stargardt disease, it is also possible that they carried disease-causing mutations in other genes or lacked a genetic disease. Traboulsi et al. previously reported that earlier disease onset and a worse BCVA were associated with an increased number of identifiable ABCA4 mutations,20 while Zahid et al demonstrated a significantly earlier disease onset in patients harbouring more than two mutations compared with those with only one detectable mutation [24]. Additionally, Rivera- Alvarez found that disease severity was related to the accumulation of multiple mutations as well as to a specific type of mutation, with null mutations the most deleterious [25]. Taken together, these results suggest that disease onset and BCVA are associated with genotype in Stargardt disease; however, none of these studies were conducted in Chinese patients.
Although the BCVA differs among groups, variations in the disease duration mean that direct comparisons are not beneficial.
The mean BCVA of eyes in the ProgStar Study was 0.88 logMAR (range, 0.66-1.0), 55% were moderately impaired (0.54-1.0 logMAR), and no eyes had a BCVA worse than 1.3 (legal blindness). The median age at symptom onset was 19 years (range, 4-64 years).13 This compares with a mean BCVA of 0.63 ± 0.44 logMAR in a study of Italian patients,14 and a mean BCVA of 0.74 ± 0.56 logMAR in the better-seeing eye and 0.87 ± 0.55 logMAR in the worse-seeing eye in a study where 86% patients were Caucasian and 11.6% were African-American. The mean age at symptom onset was 30 ± 16 years (range, 6-78 years) in this study [20]. A recent analysis of patients with Stargardt disease from the UK revealed a mean age at symptom onset of 9.6 ± 3.4 years for childhood-onset disease and 28.3 ± 7.8 years for adult-onset disease, with a mean BCVA of 0.68 ± 0.34 logMAR [11]. BCVA baseline values and age at symptom onset differ completely in a Chinese cohort. For example, we observed a worse BCVA in our cohort (1.16 ± 0.61 logMAR), 39.6% of eyes were legally blind (≤1.3 logMAR), 24.0% had a severe visual impairment (1-1.3 logMAR), and only 8.6% had a BCVA better than 0.52 logMAR. This could be explained by differences in ethnicities, genetic backgrounds, or age and disease duration. The mean age at symptom onset in our cohort (13.63 ± 12.72 years) was younger than reported in previous studies [11,17,20], which is consistent with the observed genotype distribution. Genotype A (17.4%) and B (42.2%) were identified in 59.6% of our cohort, compared with 40.9% in the ProgStar Study (A, 3.1%; B, 37.8%), and 53.5% in childhood-onset patients and 36.8% in adult-onset patients of the UK study [11]. These data suggest that Chinese patients with Stargardt disease harbour more severe genetic variants, which likely explains their worse BCVA and earlier age at symptom onset.
Our study had a number of limitations. First, it was a retrospective cross-sectional study, so further prospective studies are needed to elucidate the natural history of disease and the complex correlations of clinical characteristics and genotype. Additionally, our study participants were all enrolled from a single tertiary referral centre, so a multicentre study should be conducted to verify our findings.
In summary, our study provides a brief overview of BCVA, age at symptom onset, and associated factors in 169 Chinese patients with a clinical and genetic diagnosis of Stargardt disease. We found that BCVA levels were significantly associated with genotype and disease duration, but had no direct correlation with age at symptom onset or patient age. Genotype was also shown to be associated with age at symptom onset, with severe genotype being associated with an earlier disease onset and worse BCVA. Our data may have implications for determining appropriate candidacy and optimal timings of intervention for emerging therapies, and can serve as a well-founded reference for genetic counseling about prognosis.
Acknowledgement
The author would like to thank all the participants and the staffs for their valuable contribution to this research.
Author Contributions
Ji-Hong Wu and Ge-Zhi Xu conceived and designed the experiments. Wei Liu, Qing Chang, Ping Xu, Ji-Hong Wu, Feng-Juan Gao, Dan-Dan Wang and Ying Huang collected the clinical samples. Feng-Juan Gao, Ji-Hong Wu, Fang-Yuan Hu and Dan-Dan Wang analyzed sequencing data. Ge-Zhi Xu, Wei Liu and Feng-Juan Gao recruited patients, performed clinical examination of patients and clinical interpretation. Feng-Juan Gao, Dan-Dan Wang, Fang-Yuan Hu and Ji-Hong Wu analyzed and interpreted the data. Feng-Juan Gao and Ji-Hong Wu drafted and revised the manuscript. All authors read and approved the manuscript.
Availability of Data and Materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study adhered to the tenets of the Declaration of Helsinki, and was approved by the Ethics Committee of the Eye and ENT Hospital of Fudan University. All patients were informed about the study and provided consent.
Financial Support
This study is supported by the National Natural Science Foundation of China (Grant NSFC81770925, 81870670). Shanghai Clinical Research Plan of SHDC (NO.SHDC2020CR2041B). Shanghai municipal science and technology major projects (2018SHZDZX05). Outstanding academic leaders in Shanghai (20XD1401100). Program for Outstanding Medical Academic Leader (2019LJ01). Aging and women’s and children’s health Special project of Shanghai Municipal Health Commission (2020YJZX0102). Shanghai clinical medical center of ocular disease (2017ZZ01020). Shanghai Committee of Science and Technology (18411965100). The Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2018PT32019).
References
- Allikmets R, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature genetics. 1997; 15: 236-246.
- Bardak H, et al. Analysis of ELOVL4 and PRPH2 genes in Turkish Stargardt disease patients. Genetics and molecular research: GMR. 2016; 15.
- Imani S, et al. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget. 2018; 9: 122-141.
- Rotenstreich Y, Fishman GA & Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003; 110: 1151-1158.
- Khan M & Cremers FPM. ABCA4-Associated Stargardt Disease. Klinische Monatsblatter fur Augenheilkunde. 2020; 237: 267-274.
- Fujinami K, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015; 122: 326-334.
- Strauss RW, et al. The Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Studies: Design and Baseline Characteristics: ProgStar Report No. 1. Ophthalmology. 2016; 123: 817-828.
- Tanna P, Strauss RW, Fujinami K & Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. The British journal of ophthalmology. 2017; 101: 25-30.
- Fakin A, et al. Phenotype and Progression of Retinal Degeneration Associated With Nullizigosity of ABCA4. Invest Ophthalmol Vis Sci. 2016; 57: 4668-4678.
- Valkenburg D, et al. Highly Variable Disease Courses in Siblings with Stargardt Disease. Ophthalmology. 2019; 126: 1712-1721.
- Georgiou M, et al. Prospective Cohort Study of Childhood-Onset Stargardt Disease: Fundus Autofluorescence Imaging, Progression, Comparison with Adult-Onset Disease, and Disease Symmetry. Am J Ophthalmol. 2020; 211: 159-175.
- Runhart EH, et al. Late-Onset Stargardt Disease Due to Mild, Deep-Intronic ABCA4 Alleles. Investigative ophthalmology & visual science. 2019; 60: 4249-4256.
- Kong X, et al. Visual Acuity Change Over 24 Months and Its Association With Foveal Phenotype and Genotype in Individuals With Stargardt Disease: ProgStar Study Report No. 10. JAMA Ophthalmol. 2018; 136: 920-928.
- Arrigo A, et al. Choroidal Patterns in Stargardt Disease: Correlations with Visual Acuity and Disease Progression. J Clin Med. 2019; 8.
- Collison FT & Fishman GA. Visual Acuity In Patients with Stargardt Disease after Age 40. Retina. 2018; 38: 2387-2394.
- Kong X, et al. Progression of Visual Acuity and Fundus Autofluorescence in Recent-Onset Stargardt Disease: ProgStar Study Report #4. Ophthalmology. Retina. 2017; 1: 514-523.
- Kong X, et al. Visual Acuity Change over 12 Months in the Prospective Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: ProgStar Report Number 6. Ophthalmology. 2017; 124: 1640-1651.
- Kong X, et al. Visual Acuity Loss and Associated Risk Factors in the Retrospective Progression of Stargardt Disease Study (ProgStar Report No. 2). Ophthalmology. 2016; 123: 1887-1897.
- Gelman R, Smith RT & Tsang SH. Diagnostic Accuracy Evaluation of Visual Acuity and Fundus Autofluorescence Macular Geographic Atrophy Area for the Discrimination of Stargardt Groups. Retina. 2016; 36: 1596-1601.
- Miraldi Utz V, et al. Predictors of visual acuity and genotype-phenotype correlates in a cohort of patients with Stargardt disease. Br J Ophthalmol. 2014; 98: 513-518.
- Kim LS & Fishman GA. Comparison of visual acuity loss in patients with different stages of Stargardt’s disease. Ophthalmology. 2006; 113: 1748- 1751.
- Fishman GA, Farber M, Patel BS & Derlacki DJ. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology. 1987; 94: 809-814.
- Simonelli F, et al. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic research. 2005; 37: 159-167.
- Zahid S, et al. Clinical phenotypes and prognostic full-field electroretinographic findings in Stargardt disease. American journal of ophthalmology. 2013; 155: 465-473.e463.
- Riveiro-Alvarez R, et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology. 2013; 120: 2332-2337.
- Alabduljalil T, et al. Correlation of Outer Retinal Degeneration and Choriocapillaris Loss in Stargardt Disease Using En Face Optical Coherence Tomography and Optical Coherence Tomography Angiography. American journal of ophthalmology. 2019; 202: 79-90.
- Strauss RW, et al. Progression of Stargardt Disease as Determined by Fundus Autofluorescence Over a 12-Month Period: ProgStar Report No. 11. JAMA ophthalmology. 2019.
- Wang DD, et al. Clinical and Genetic Characteristics of Chinese Patients with Occult Macular Dystrophy. Invest Ophthalmol Vis Sci. 2020; 61: 10.
- Hu FY, et al. ABCA4 Gene Screening in a Chinese Cohort With Stargardt Disease: Identification of 37 Novel Variants. Frontiers in genetics. 2019; 10: 773.
- Gao FJ, et al. Mutation spectrum of the bestrophin-1 gene in a large Chinese cohort with bestrophinopathy. Br J Ophthalmol. 2019.
- Gao FJ, et al. Genetic and clinical findings in a large cohort of Chinese patients with suspected retinitis pigmentosa. Ophthalmology. 2019.
- Gao FJ, et al. Expanding the clinical and genetic spectrum of Heimler syndrome. Orphanet J Rare Dis. 2019; 14: 290.
- Lange C, Feltgen N, Junker B, Schulze-Bonsel K & Bach M. Resolving the clinical acuity categories “hand motion” and “counting fingers” using the Freiburg Visual Acuity Test (FrACT). Graefe’s archive for clinical and experimental ophthalmology. 2009; 247: 137-142.
- Holladay JT. Proper method for calculating average visual acuity. Journal of refractive surgery. 1997; 13: 388-391.