
Letter to Editor
J Ophthalmol & Vis Sci. 2024; 9(1): 1090.
Wolfram Syndrome: A Case Report and Review of the Literature
Hasnaoui Ihssan*; Hazil Zahira; Salma Hassina; Krichen Amine; Y Akkenour; L Serghini; Abdellah Elhassan
Department of Ophthalmology B, Faculty of Medicine and Pharmacy, Hospital of Specialities, CHU Ibn Sina Rabat, University Mohamed V, Morocco
*Corresponding author: Hasnaoui I Department of Ophtalmology B, Faculty of Medicine and Pharmacy, University Mohamed V, Av. Abderrahim Bouabid, 10100 Rabat, Morocco. Email: ihssanhasnaoui@gmail.com
Received: March 07, 2024 Accepted: April 18, 2024 Published: April 25, 2024
Introduction
First described by Wolfram in 1938, Wolfram Syndrome (WS) is a rare neurodegenerative disease; its acronym DIDMOAD groups together the main clinical features of the syndrome: juvenile-onset diabetes mellitus followed later by optic atrophy leading to blindness, diabetes insipidus, hearing loss and other neurological, urinary and endocrine dysfunctions. Through this case and a review of the literature, we recall the main clinical signs of WS.
Case Report
A 15-year-old girl from a consanguineous marriage, with a history of congenital heart disease (atrial septal defect), type I diabetes since the age of 9 and dibetus insipidus, consulted for a progressive and profound decline in visual acuity that began 3 years ago.
The ophthalmological examination revealed reduced visual acuity of one metre in both eyes, and isolated bilateral optic atrophy on the fundus (Figure 1). The rest of the ophthalmological examination was unremarkable.
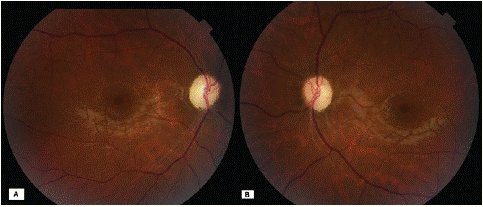
Figure 1: Retinography of the right eye (A) and left eye (B) showing total papillary pallor.
In view of the clinical picture, an ENT opinion with an audiogram was requested; the results were in favour of a sensorineural hearing loss in the left ear; SW was suspected in the patient, a genetic study was carried out and confirmed a homozygous WFS1-528Y/D mutation.
Discussion
Wolfram syndrome is a rare clinical entity characterized by clinical and genetic polymorphism; two responsible genes (WFS1 and WFS2) have been identified, and the disease is inherited in an autosomal recessive mode, although autosomal dominant mutations have been described.
The major signs of this syndrome, diabetes mellitus and optic atrophy, appear in childhood and the picture is then progressively completed by: hearing impairment, which is generally diagnosed in the 2nd or 3rd decade of life, probably following progressive deterioration of the central nervous system; neurological complications affect 62% of patients, appearing at an average age of 16 years [1]. Cerebellar ataxia is the most common neurological complication; other signs include dysphagia, dysarthria, areflexia and epilepsy. Urinary tract problems are frequently reported, with neurogenic bladder and its complications affecting up to 90% of patients. Other signs include severe depression, psychosis, sleep disorders, gastrointestinal problems, etc.
The prognosis is poor, with death occurring at the median age of 39, the major cause being respiratory failure following brainstem atrophy and neurodegeneration.
The history and clinical manifestations, based on the presence of optic nerve atrophy following the diagnosis of diabetes mellitus before the age of 15, should arouse the clinician's suspicion. However, the visual abnormalities accompanying diabetes in patients with SW may lead to a misdiagnosis of type 1 diabetes with diabetic retinopathy in some children and adolescents, which may result in a delayed revelation of this diagnosis and an underestimation of the prevalence of WS in the childhood population [2].
Conclusion
In SW, every organ and system can be affected by neuregenerescence, hence the importance of regular monitoring in a multidisciplinary centre; in this way, knowledge of the different clinical signs and their chronology of appearance means that the patient with WS can be given the right information for reintegration and appropriate social guidance.
Author Statements
Conflicts of Interest
The authors have no conflicts of interest to declare.
Consent
Written informed consent was obtained from the patient and his parents for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
References