
Case Report
Austin J Orthopade & Rheumatol. 2024 ; 11(2) : 1131.
Extraosseous Ewing’s Sarcoma - A Case Report
El Maqrout Amine1*; Rhouri F, Mekkaoui J1; Boufettal M2; Bassir RA2; Kharmaz M1; Lamrani MO1; Berrada MS1
¹Department of Trauma and Orthopedic Surgery, Ibn Sina University Hospital, Faculty of Medicine, Mohammed V University of Rabat, Rabat, Morocco
²Department of Anatomy, Faculty of Medicine, Mohammed V University of Rabat, Rabat, Morocco
*Corresponding author: EL MAQROUT AMINE Department of Trauma and Orthopedic Surgery, Ibn Sina University Hospital, Faculty of Medicine, Mohammed V University of Rabat, Rabat, Morocco. Email: amineelmaqrout96@gmail.com
Received: June 27, 2024 Accepted: July 10, 2024 Published: July 17, 2024
Abstract
Early detection and aggressive treatment are crucial for improving the prognosis of patients with extraosseous Ewing’s sarcoma. This case illustrates the successful management of a patient with this rare disease, highlighting the importance of multidisciplinary healthcare teams in providing optimal care. Continued research and collaboration are essential in advancing the understanding and treatment of Ewing’s sarcoma to enhance outcomes for affected individuals.
Introduction
Ewing's sarcoma is a rare form of cancer that develops in soft tissues or bones. In cases of extraosseous Ewing's sarcoma, the tumor forms outside the bones, making it even more rare. This disease mainly affects young adults and can be challenging to diagnose due to its rarity.
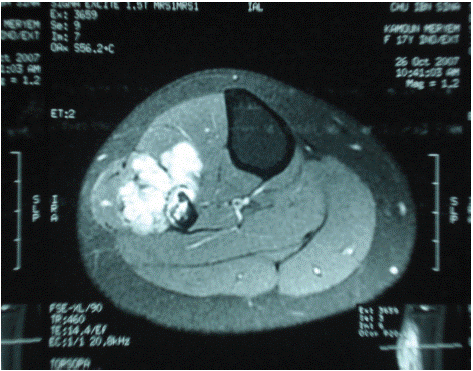
Figure 1: MRI of the right leg: tumor on the lateral side of the leg.

Figure 2: MRI of the right leg.
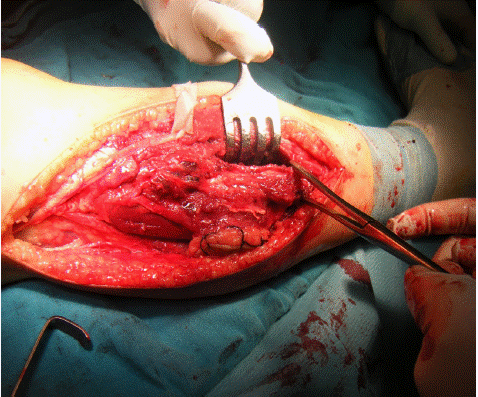
Figure 3: Ewing’s sarcoma.
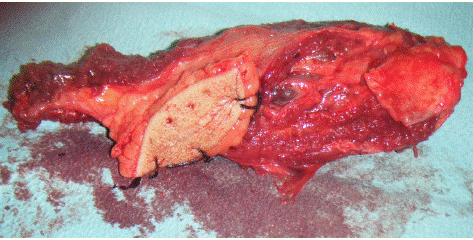
Figure 4: Surgical specimen.
In this article, we present the case of a patient diagnosed with extraosseous Ewing's sarcoma in her right leg.
The patient's treatment involved a combination of surgery to remove the tumor, chemotherapy, and radiotherapy to eliminate any remaining cancer cells. Thanks to prompt and effective management, the patient was able to recover and currently shows no signs of recurrence.
This case highlights the importance of early detection and aggressive treatment of extraosseous Ewing's sarcoma to improve chances of recovery. It also emphasizes the significance of close collaboration between healthcare professionals to ensure the best possible prognosis for patients with this rare and aggressive disease.
Observation
This is a 33-year-old patient with no significant medical history, presenting for the past two and a half years with a rapidly evolving, painful swelling near the proximal right tibia, without any inflammatory signs.
Clinical examination revealed a large swelling in the upper third of the right leg. The knee joint was normal.
The rest of the clinical examination was unremarkable, with no palpable lymph nodes.
Laboratory tests were normal.
X-rays showed an opacity near the upper third of the right tibia.
Further evaluation with MRI revealed a heterogeneous tumor process with hyperintensity occupying the upper third of the right leg. A biopsy was performed, confirming the diagnosis of Ewing's sarcoma. Staging studies were negative. The patient received an initial combination chemotherapy regimen with cyclophosphamide and doxorubicin to reduce the size of the tumor, and two months later, underwent tumor resection.
Discussion
Ewing's sarcoma is a rare form of tumor that primarily affects young adults, both men and women [1].
Pain is often the most common symptom and does not respond to typical analgesics, with swelling typically present [1-3].
Radiological examinations are essential for an accurate diagnosis, with current preference for MRI over CT scans [3], as it allows better characterization of tumor elements and their environment.
The definitive diagnosis relies on a biopsy. Macroscopically, the tumor is often whitish, multi-lobed, friable, infiltrating and destroying all layers of a region. Microscopically, it consists of small cells resembling other sarcomas with a mesenchymal ultrastructure [4]. Prognostic factors include tumor volume, initial site, patient age, presence of metastases at diagnosis, and response to chemotherapy.
Treatment primarily involves chemotherapy to achieve tumor regression, based on combination chemotherapy to enhance effects without increasing toxicity. With the use of intensive chemotherapy, the 5-year survival rate is around 50 to 60% [5,6].
Surgery and radiotherapy may also be necessary depending on the situation.
Monitoring patients and metastasis surveillance remain topics of debate in medical literature. The prognosis is often reserved, with a risk of local recurrence or long-term metastases [6].
Conclusion
Ewing's sarcoma is a malignant tumor that primarily develops in bone tissue, but can rarely affect soft tissues. The diagnosis is often complex, even with detailed anatomopathological analyses. Treatment typically involves a combination of surgery, radiotherapy, and chemotherapy.
Patient follow-up requires regular clinical and radiological surveillance.
References
- Delatre O, Dauohinot L. famille des tumeurs d’Ewing, MTP. 1998: 2.
- Pritchard DJ, Dahlin DC, Dauphine RT. Ewing’s Sarcoma. J Bone Joint Surg. 1074; 57: 10.
- Fizazi K, Spielmenn M, Le Cesne. A Sarcome d’Ewing et tumeurs neuro-ectodermiques primitives (PNET) cancérologie aujourd’hui. 1996; 5: 16-25.
- Stines J. Métastases pulmonaires et bronchiques. Encycl Méd Chir (Elseveir Paris), radiodiagnostic. Coeur-Poumon. 1996; 32-461-A-10.
- Simon MA, Enneking WF. The management of soft tissue sarcomas of the extremities J. Bone Joint Surg. 1976; 58: 317-27.
- Sudanese A, Toni A, Ciaroni D, Bacci G, Picci P, Barbieri E, et al. The role of surgerical therapy in patients with nonmetatasic Ewing’s sarcoma of the limbs. Clinical orthopaedics and related research. 1993; 286: 225-240.