
Case Report
Austin J Orthopade & Rheumatol. 2016; 3(3): 1038.
A Sinister Crisis in Scleroderma
Elizabeth H Federici¹, Nancy J Olsen¹, Michael G Bayerl² and Eileen F Hennrikus¹*
¹Department of Medicine, Penn State Hershey Medical Center, USA
²Department of Pathology, Penn State Hershey Medical Center, USAzil
*Corresponding author: Eileen F Hennrikus, Department of Medicine, Penn State Hershey Medical Center, Pennsylvania, USA
Received: August 11, 2016; Accepted: September 16, 2016; Published: September 19, 2016
Abstract
We describe a presentation of Systemic Sclerosis (SSc) with rapid development of Scleroderma Renal Crisis (SRC) and multiorgan failure leading to death. Diagnoses were confirmed on post-mortem examination. SSc usually presents with skin thickening of the fingers and the presence of Anti-Nuclear Antibodies (ANA).Other associated clinical findings include fingertip lesions, telangiectasia, abnormal nailfold capillaries, pulmonary arterial hypertension, interstitial lung disease, and Raynaud’s phenomenon. ANA testing is negative in only a small minority of patients. Scleroderma Renal Crisis is characterized by acute renal failure, hypertension, and microangiopathic hemolytic anemia with thrombocytopenia, but occurs without hypertension in some cases. This case demonstrates an atypical case of SSc and SRC in that ANA testing was negative and Scleroderma Renal Crisis developed without hypertension. This presentation illustrates the challenge in diagnosing and managing SSc and SRC in a critically ill patient.
Keywords: Systemic Sclerosis; Scleroderma renal crisis; Anti-nuclear antibodies
Introduction
Systemic Sclerosis (SSc) is an autoimmune disease in which autoantibodies against cellular antigens exist alongside diffuse fibrosis and vascular changes of the skin and internal organs. SSc most commonly involves the kidneys, esophagus, heart, and lungs, although nearly all internal organs may be affected [1]. Scleroderma associated renal failure was first reported in 1863 but was not recognized as a complication of SSc until described in a case series by Moore and Sheehan in 1952 [2]. Scleroderma Renal Crisis (SRC) is typically characterized by accelerated hypertension and acute renal failure, but occurs without hypertension in 10% of cases [3]. Microangiopathic hemolytic anemia and thrombocytopenia are recognized in approximately half of cases of SRC [4]. This report describes a man who presented with a suspected diagnosis of Systemic Sclerosis and developed normotensive Scleroderma Renal Crisis during hospitalization.
Case Presentation
A 56-year-old African American man was admitted with recurrent episodes of abdominal distention, nausea and vomiting. On initial examination he had skin thickening of the fingers of both hands extending proximally beyond the metacarpophalangeal joints to the upper arms and skin thickening of the toes, feet, and ankles. A small bowel follow-through demonstrated mild bowel dilation and marked delayed transit of contrast, but no evidence of mechanical obstruction. There was a “hide-bound” or “stacked coin” appearance of the bowel (Figure 1).
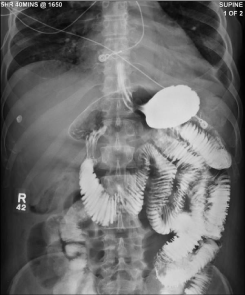
Figure 1: Small bowel follow-through.
He had a recent history of dry cough and myalgias treated with 20 mg prednisone daily and a brief course of methotrexate for an unspecified rheumatologic disorder. Two weeks prior to presentation, a high resolution CT chest demonstrated ground glass opacities and peripheral reticulations consistent with interstitial lung disease (Figure 2).
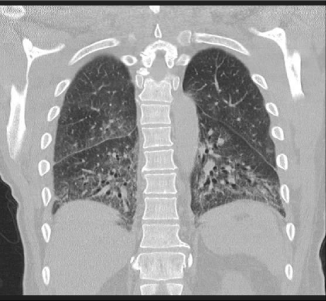
Figure 2: CT of the chest in the coronal plane.
His medical history also included gastroesophageal reflux disease and hypertension. He did not complain of Raynaud symptoms. His other medications were pantoprazole, hydrochlorothiazide, and amlodipine. He worked as an administrator and had no known exposures to toxins.
Based on this presentation, a diagnosis of Systemic Sclerosis (SSc) was suspected. His acute illness was attributed to intestinal pseudoobstruction secondary to SSc and was managed with bowel rest, intravenous rehydration, and nasogastric tube to suction. Six days later, scleral icterus was noted on examination.
On hospital day six, the patient’s temperature was 37.2°C, heart rate 75 beats per minute, blood pressure 122/82 mm Hg, respiratory rate 24 breaths per minute, and oxygen saturation 96% on 4L oxygen via nasal cannula. The patient appeared fatigued. He was intermittently confused but oriented to person, place, and time. The sclerae were icteric. Cardiac examination was normal. Lung examination revealed tachypnea, coarse breath sounds and bibasilar crackles. Bowel sounds were hypoactive. The abdomen was distended, soft and non-tender. Skin examination was unchanged from admission.
The patient’s laboratory values on presentation and hospital day six are in Table 1.
![]()
Reference Range
On Presentation
Hospital Day 6
White-cell count (per μL)
4,000 - 10,400
11,550
12,240
Neutrophils (%)
40 - 70
88.9
88.6
Lymphocytes (%)
22 - 44
4.8
4.7
Monocytes (%)
4 - 11
4.8
5.8
Eosinophils (%)
0 - 8
1.0
0.1
Basophils (%)
0 - 3
0.2
0.1
Hematocrit (%)
39 - 48
38.4
31.7
Hemoglobin (g/dL)
13 - 17
13
10.3
Platelet count (per μL)
150 - 350
346,000
94,000
Total bilirubin (mg/dL)
0.2 - 1.3
1.6
4.7
Direct bilirubin (mg/dL)
0 - 0.6
0.7
1.6
Sodium (mmol/L)
137 - 145
134
142
Urea nitrogen (mg/dL)
7 - 20
30
45
Creatinine (mg/dL)
0.7 - 1.3
0.76
1.07
Aspartate aminotransferase (per L)
15 - 46
43
68
Alanine aminotransferase (per L)
13 - 69
34
41
Alkaline phosphatase (per L)
38 - 126
129
87
Erythrocyte sedimentation rate (mm/hour)
0 - 25
67
Complement C3 (mg/dL)
88 - 165
109
Complement C4 (mg/dL)
14 - 44
29
Protime (seconds)
12 - 14.2
14.8
International normalized ratio
0.87 - 1.09
1.15
Prothrombin time (seconds)
23 - 35
32
Fibrinogen (mg/dL)
208 - 435
338
Fibrin degradation product (μg/mL)
Negative
=20
Lactate dehydrogenase (per L)
313 - 618
1,212
Haptoglobin (mg/dL)
43 - 212
<7
Table 1: Laboratory values.
Direct antiglobulin test was negative. Peripheral blood smear demonstrated 1 to 2 schistocytes per high power field and spherocytes.
Serological studies including Rheumatoid Factor (RF), anti-Cyclic Citrullinated Peptide (anti-CCP), Anti-Nuclear Antibody (ANA) screen by Immunofluorescent Assay (our ANA assay uses HEp2-A as the substrate), Anti-Topoisomerase I (anti-Scl-70), Anticentromere (ACA), anti-RNP antibodies, Anti-Neutrophil Cytoplasmic Antibody (ANCA) and antiphospolipid antibodies were negative.The patient became progressively encephalopathic, dyspneic, hypoxic, and anuric but remained normotensive. Creatinine increased to 3.4 milligrams per deciliter. On hospital day eight, he was endotracheally intubated and mechanically ventilated for acute respiratory failure and renal replacement therapy was initiated. A skin biopsy demonstrated sclerosing dermatitis including sclerotic collagen bundles within the dermis, loss of fat around eccrine glands, and decreased number of CD34 cells.
Diagnosis
This patient’s characteristic skin findings on examination and biopsy, interstitial lung disease and recurrent intestinal pseudoobstruction were consistent with a diagnosis of SSc. The subsequent development of hemolytic anemia, thrombocytopenia, and acute renal failure suggested the presence of a thrombotic microangiopathy. The suspected diagnosis was Scleroderma Renal Crisis (SRC). Differential diagnoses included thrombotic Thrombocytopenic Purpura (TTP) and complement-mediated hemolytic uremic syndrome.
Diagnosis of SSc is based on clinical findings and is supported by the presence of SSc-associated autoantibodies. Classification criteria published by the American College of Rheumatology and European League against Rheumatism define any patient who has “skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints” as sufficient for classification as having SSc. Weighted points are assigned for other findings, including, puffy fingers, sclerodactyly, digital tip ulcers, fingertip pitting scars, telangiectasia, abnormal nailfold capillaries, pulmonary arterial hypertension, interstitial lung disease, Raynaud’s phenomenon, and SSc-associated antibodies (anti-centromere, anti-topoisomerase I, and anti-RNA polymerase III) [5].
There are no classification criteria for SRC and the diagnosis is made clinically, based on a previous diagnosis of SSc, acute renal failure, hypertension, and microangiopathic hemolytic anemia. Renal biopsy may be done to confirm the diagnosis. Characteristic histologic findings include occlusive microvascular thrombosis, fibrinoid necrosis, onion-skin lesions, and fibrointimal sclerosis [6].
Management
The patient was treated for SRC with an angiotensin converting enzyme-inhibitor, but this was stopped after several days due to profound hypotension. Plasma exchange was initiated, but TTP was eventually excluded as a diagnosis based on lack of response in platelet count to this intervention and results of an ADAMTS13 activity assay of 45% (less than 10% is considered consistent with TTP). Treatment for possible complement-mediated hemolytic uremic syndrome was also started with once weekly dosing of eculizumab, but later results indicated normal levels of complement factors H, I, and B, making this diagnosis unlikely. Despite continued supportive therapy, on hospital day nineteen, the patient developed sustained ventricular tachycardia and later pulseless electrical activity with cardiac arrest and expired.
Postmortem examination limited to the chest, abdomen, and pelvis was obtained. Histopathological examination of the lungs demonstrated bilateral diffuse fibrosis consistent with nonspecific interstitial pneumonitis with foci of alveolar hemorrhage. Examination of the kidneys revealed bilateral diffuse cortical coagulative necrosis and fibrin thrombi within the arterioles, and capillary loops consistent with SRC (Figure 3).
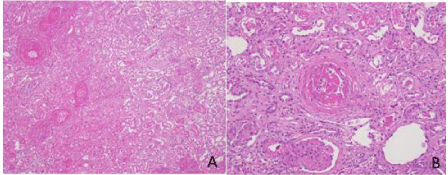
Figure 3: Postmortem histopathological examination of the kidney.
A, Kidney, extensive coagulative necrosis and a laminated thromboembolus
(upper left) [H&E stain, 100X magnification].
B, Kidney, viable cortex with an organizing thromboembolus and characteristic
onion-skin changes in arterioles [H&E stain, 400X magnification].
Discussion
The final diagnoses were acute thrombotic microangiopathy and normotensive SRC, based on both the clinical presentation and findings on postmortem examination.
Recent moderate to high dose corticosteroid use is a known risk factor for the development of SRC although a direct causal role has not been demonstrated [4]. The pathogenesis of SRC is thought to be secondary to renin-mediated malignant hypertension. Intimal thickening of the renal interlobular and arcuate arteries occurs as a result of endothelial cell injury. Decreased renal perfusion from narrowed arterial vessels results in hyperplasia of the juxtaglomerular apparatus and increased renin activity [7]. Vascular endothelial abnormalities from fibrin deposition result in impaired circulation and physical trauma to red blood cell membranes, resulting in microangiopathic hemolytic anemia [3].
This patient did not have typical SSc-associated antibodies, nor was he ever hypertensive during his final hospitalization. However, he did have characteristic physical examination findings, classic multiorgan involvement and histologic changes consistent with SSc. The patient did have an identifiable risk for SRC with his recent exposure to a moderate dose of prednisone. Population studies have identified a small subgroup of SSc patients who were ANA negative. These patients were more commonly male [8].
There are no disease-modifying drugs for the treatment of SSc. Therapy remains limited to management of organ-specific complications. SSc has high morbidity and is frequently fatal [9,10]. In a prospective study, 10-year survival rates ranged from 54% to 66% [10]. The majority of morbidity and mortality of SSc arises from visceral organ-based complications [9], especially pulmonary involvement. Previously, SRC was the primary cause of SSc-related deaths [10], but with the introduction of angiotensin converting enzyme-inhibitors, modern dialysis techniques, and overall improved patient care, mortality from this cause has decreased [3,4,7,10]. This case illustrates how SSc can smolder along as a slowly progressive and poorly defined or undetected condition and then abruptly flare into a rapidly progressive, deadly disorder with microangiopathic hemolytic anemia and multiorgan system failure. It also highlights the challenge in diagnosing SSc and SRC in a critically ill patient. In cases such as this one, a diagnosis of SRC may only be confirmed after treating for and excluding other diagnoses.
References
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009; 360: 1989-2003.
- Rodnan GP, Benedek TG. An historical account of the study of progressive systemic sclerosis (diffuse scleroderma). Ann Intern Med. 1962; 57: 305-319.
- Helfrich DJ, Banner B, Steen VD, Medsger TA Jr. Normotensive renal failure in systemic sclerosis. Arthritis Rheum. 1989; 32: 1128-1134.
- Penn H, Howie AJ, Kingdon EJ, Bunn CC, Stratton RJ, Black CM, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM. 2007; 100: 485-494.
- van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Ann Rheum Dis. 2013; 65: 2737-2747.
- Batal I, Domsic RT, Medsger TA, et al. Scleroderma Renal Crisis: A Pathology Perspective. Int J Rheumatol. 2010; 2010: 543704.
- Denton CP, Lapadula G, Mouthon L, Müller-Ladner U. Renal complications and scleroderma renal crisis. Rheumatology (Oxford). 2009; 48; 32-35.
- Salazar GA, Assassi S, Wigley F, Hummers L, Varga J, Hinchcliff M, et al. Antinuclear antibody-negative systemic sclerosis. Semin Arthritis Rheum. 2015. 44: 680-686.
- Khanna D, Denton CP. Evidence-based management of rapidly progressing systemic sclerosis. Best Pract Res ClinRheumatol. 2010; 24: 387-400.
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis 2007; 66: 940-944.