
Case Report
Austin J Pathol Lab Med. 2019; 6(1): 1022.
Novel Hepatosplenic T-Cell Lymphoma in Young Adult with Sickle Cell Disease
Ghazal MA1* and ALHajjaj D2
1Department of Hematopathology, Dammam Regional Laboratory, Saudi Arabia
2Department of Medicine, Dammam Complex Hospital, MOH, KSA
*Corresponding author: Ghazal MA, Department of Hematopathology, Dammam Regional Laboratory, Saudi Arabia
Received: December 20, 2018; Accepted: January 30, 2019; Published: February 06, 2019
Abstract
Hepatospenic T-cell Lymphoma (HSTL) is rare & an aggressive type of extranodal lymphoma, the disease represent 1-2% of all peripheral T-cell lymphoma [1]. Its incidence might be underestimated, because the disease may mimic other conditions & the diagnosis is sometimes difficult to be established .It has been seen in patients receiving long-term immunosuppressive therapy, following AML or EBV positive lymphoproliferative disorders or during pregnancy. Several cases were reported recently in patients with crohns disease treated with azathioprine & anti-tumor necrosis factor agent infliximab. We present here peculiar scenario of young male adult known case of sickle cell disease patient whose been diagnosed with gammadelta-T-cell lymphoma, after quite long history of unexplained organomegaly & fever.
Introduction
HSTL is rare, and the world health organization prefers the term hepatosplenic T-cell lymphoma in the current WHO classification of tumors of haematopoietic & lymphoid tissues. The disease occurs mainly in young adults presenting with hepatomegaly, splenomegaly but without lymphadenopathy. Most patients have B-symptoms & cytopenias. In fact bone marrow involvement is nearly always present. Neoplastic cells infiltrate & expand the bone marrow sinuses, a feature that is highly characteristic & thus a useful diagnostic criterion, irrespective of their gammadelta or alphabeta phenotype, HSTL shows a clonal rearrangement of TCRG gene. Recently gene expression profiling studies have shown that the HSTL signature was distinct & characterized by over expression of gene encoding KIRs molecules. In conventional cytogenetic & FISH studies, 50-80% were characterized by the presence of an isochromsome 7q(i[7][q10]) this is occasionally the sole karyotypic abnormality suggesting that it plays a role in pathogenesis, in addition to trisomy 8 & loss of chromosome Y. The major differential diagnoses include aggressive NK-cell lymphoma/leukemia, T-Cell large granular lymphocytic leukemia & other gammadelta T-cell lymphomas.
Case Presentation
26 years old young male, known case of SCD on hydroxyurea, with history of multiple transfusion, was admitted on 19 September 2018 because of very high white blood cell count 91,000/ul with abnormal liver function test. Going back to his history, patient was admitted to another hospital 3 months back with abdominal pain, diagnosed as sequestration crisis at that time followed by splenctomy, postoperatively was found to have high grade fever reaching up to 40°C, CT abdomen was done & showed collection at site of operation, so explored & the patient was kept on antibiotics, but unfortunately he was continued to have fever, so bone marrow was done but was inadequate, however trephine biopsy showed grad III fibrosis & repetition was suggested. After splenctomy the patient decided to stop hydroxyurea, since then his white blood cells started to rise up. The patient gave history of significant weight loss (around 10KG). all septic work up done were negative.
On clinical examination, he was jaundiced, thin, tall, pale with fever of round 40°C. chest examination was clear while abdomen shows long longitudinal scar with huge hepatomegaly. There is daily increase in WBC counts, although he is covered with imipinem & hydroxyurea. Peripheral blood examination shows total white blood count of around 147,870/cumm. Hb-6.9 g/dl, Platelets-1340000/cumm next day total white cells rise to 161,320/cumm. While both hemoglobin & platelets counts dropped down to Hb-6.7 g/dl, & Platelets-930000/ cumm, PT &APTT are normal, however total bilurbin is elevated 22.11mg/dl(normal is less than 0.3mg/dl), mainly direct 16.4mg/dl(normal range is 0.3-1.0mg/dl), AST (GOT):381(normal is 10-40 units/L)., ALT(GPT):105(normal is 7-56 units/l).GGT:194U/L(15-85) LDH:2508U/L(normal range is (100-190), alkaline phosphastase is 427U/L(46-116).
Peripheral blood morphology examination shows Marked Leukocytosis, Leukoerythroblastic reaction, Nucleated RBC- 47/100 WBC. There is infiltration by around (75%) atypical lymphoid cells Majority are medium sized have moderate to abundant amount of cytoplasm some of them have round to oval nuclei with few granules resembling LGL cells, Others have mostly oval nuclei with open chromatin, one to two nucleoli (blast like morphology), (Figure 1).
Flow cytometry was conducted on peripheral blood and shows that majority of population are T-Lymphocytes (80%) Double Negative for CD4, CD8 and Positive for s-CD3, c-CD3, CD2, CD7, CD16, CD56, CD11b, CD11c & express gamma delta-T-cells receptor. They are Negative for CD34, TdT, HLA-DR, CD117, CD13, CD33, CD14, CD64, CD4, CD5, CD8, CD10, CD19, CD22, CD99, CD1a, CD25, CD20, CD79a, CD57, Alpha Beta and c-MPO, (Figure 2). Bone marrow studies were conducted, unfortunately was dry tap (both sides are tried), however bone Marrow Trephine Biopsy was adequate with increased cellularity of around 80-90% showing interstitial infiltration by variable sized lymphocytes, majority are small size with clumped chromatin along with moderate size lymphocytes with slightly open chromatin & prominent nucleoli. Myeloid series are suppressed while erythroid & megakaryocytes are present, (Figure 3).
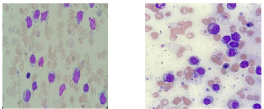
Figure 1: Infiltration by atypical lymphocytes with blasts like morphology.
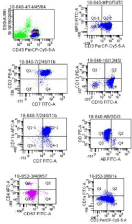
Figure 2: Flow cytometry from peripheral blood, showing gamma delta
T-cells expression in the T lymphocytes with aberrant CD11b.
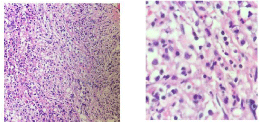
Figure 3: Interstitial infiltration by atypical lymphocytes.
Reticulin Stain
Grade III Positive Reticulin stain. (WHO 2016 grading), (Figure 4) while immunohistochemical studies with CD3, highlights increased positive T-lymphocytes with marked interstitial infiltrations (Figure 5), however TDT, CD34 & CD57 are negative.
CT scan of abdomen & pelvis shows that the liver is massively enlarged 30.0 cm, with no focal lesion. Gallbladder is surgically removed while the spleen is not seen (postsplenctomy). Both adrenal, and pancrease are unremarkable.no size significant lymph node .the visualized dorsolumbar spine demonstrate SCD changes. chest & neck CT scan are unremarkable with no evidence for size significant lymph node. In our case, cytogenetic examination failed to yield metaphase; while FISH study using LSI7q probe shows 3 signals of chromosome 7 in 41% of interphase nuclei examined, (Figure 6). However, after diagnosis was made the patient went to tertiary hospital where molecular confirmation was made & gamma delta t cell receptor gene rearrangement was confirmed eventually the patient started on chemotherapy (hyper CVAD protocol) & in search for HLA matched donor for future HSCT (Hematopoietic Stem Cell Transplantation).

Figure 4: Grade III reticulin fibrosis.

Figure 5: Immunohistochemistry with CD3 highlighting the T-lymphocytes.
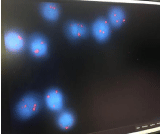
Figure 6: Fish study performed using a probe LSI7q (telomere end) shows 3
signals for 7q in 41% of the interphase nuclei examined (82/200).
Discussion
In this case report we present a case of hepatosplenic T- cell lymphoma emerging from a young adult male with sickle cell disease. We are not sure whether the diagnosis was delayed or not, this is because he had almost 3 months history of B-symptoms & huge liver & spleen, in which he thought to have splenic sequestration & accordingly splenectomy was done 2 months prior to presenting to our hospital.
Strangely histopathological examination with extensive immunohistochemistry did not show any evidence for infiltrations in the spleen, while bone marrow studies at that time were not adequate also, so presenting with leukemic phase in such T-cell lymphoma although uncommon is most probably due to absence of spleen. Although the patient presented to us with marked leukocytosis accompanied by daily increase in the count along with un resolving fever, initially the phenotype conducted from peripheral blood by flowcytometry shows expansion of gammadelta T-cells population with aberrant CD11b & CD11c,but still we needed more confirmation from bone marrow studies, unfortunately it was dry tap due to heavy infiltration as evident in the bone marrow trephine biopsy the challenge is the differential diagnosis which include aggressive NK-cell lymphoma/leukemia, T-Cell large granular lymphocytic leukemia, since gammadelta T-cell receptor is expressed, we did not consider NK-cell lmphoma/leukemia & on the other hand immunophenotype for CD57 is negative by both flowcytometry & immunohistochemistry eventually HSTL is considerd the most likely diagnosis, then molecular studies shows gammadelta T-cell receptor gene rearrangement which confirmed the clonality .
Sickle Cell Disease (SCD) are so common in our area (east province, kingdom of Saudi Arabia). In our region the clinical & laboratory features like splenomegaly & high hbF are the predominant and or coexistent of alpha thalassemia trait or hereditary persistence of HB-F [2]. Sickle hepatopathy is an umbrella term, encompassing diverse hepatic pathology arising from a variety of insults to the liver which can occur in patients with sickle cell anemia. It occurs predominantly in patients with homozygous HbSS disease. The spectrum of clinical phenotypes seen include asymptomatic abnormality of liver function, life-threatening acute liver disease, end-stage cirrhosis & hematological malignancies such as acute leukemias, chronic lymphocytic leukemia & multiple myeloma [3].
we think that our patient is rare case of hepatosplenic T-cell lymphoma in sickle cell disease homozygous (HBSS), An important question that remains unanswered is why this patient developed HSTL, is there trigger factor in sickle cell disease due to continuous insult, if so is it necessary to consider such diagnosis in all sickle cell disease patients & whats the underlying pathophysiology. Since the genetic signature in HSTL is characterized by over expression of genes encoding KIRs molecules that is making it different from other T-cell lymphoma. KIR expression is induced by chronic antigenic stimulation, & may be related to the underlying pathogenesis of the disease.another question that should be thought of is weather hydroxyurea might be a possible cause for this SCD patient to develop HSTL as a possible rare side effect, this question can be answered with continuous monitoring & reporting of all possible side effects of this drug in sickle cell patients.
Conclusion
HSTCL is a rare disease, though more commonly seen in young adolescent males. Around 20% of these young patients have a history of immunosuppression in the form of treatment of malignancy (Hodgkin lymphoma or acute myeloid leukemia), use of infliximab in inflammatory bowel disease, or solid organ transplantation, [4] being sickle cell disease weather it is coincidental diagnosis or an outcome of continuous irritation & insult to the liver or a possible rare side effect of hydroxyurea needs further complicated studies by gene expression profiling studies for further understanding.
References
- Daniel EJA, Arber DA, Campo E, Harris NL, Quintanilla-Fend L. Hematopathology, 2nd Edition. chapter 34: 631.
- Alabdulaali MK. Sickle cell disease patients in eastern province of Saudi Arabia suffer less severe acute chest syndrome than patients with African haplotypes. Thorac Med. 2007; 2: 158-162.
- Tavabie OMRCP, Suddle AR, FRCP. Lymphoma and Hematological Conditions: II. Sickle Cell Hepatopathy and Other Hematological Diseases. Clinical Liver Disease. 2016; 8.
- Sharma S, Sreedharanunni S, Sachdeva SUPM, Prakash G, Varma N. Leukemic phase of hepatosplenic T-cell lymphoma: A great masquerader of acute leukemia. Indian journal of pathology & microbiology. 2017; 60: 437-438.