
Case Report
J Pediatri Endocrinol. 2016; 1(1): 1004.
ACTH-Independent Cushing Syndrome in an Adolescent Girl due to Micronodular Adrenal Hyperplasia
Cebeci AN¹*, Guven A², Mutus M³ and Zemheri E4
¹Derince Training and Research Hospital, Clinic of Pediatric Endocrinology, Kocaeli, Turkey
²Amasya University Medical Faculty, Department of Pediatric Endocrinology & Goztepe Training and Research Hospital Pediatric Endocrinology Clinic, Istanbul, Turkey
³Goztepe Training and Research Hospital, Clinic of Pediatric Surgery, Istanbul, Turkey
4Goztepe Training and Research Hospital, Clinic of Pathology, Istanbul, Turkey
*Corresponding author: Ayse Nurcan Cebeci, Derince Training and Research Hospital, Clinic of Pediatric Endocrinology, Kocaeli, Turkey
Received: June 09, 2016; Accepted: June 13, 2016; Published: August 16, 2016
Abstract
Objective: Cushing Syndrome (CS) has a bimodal age distribution in children; preschool children mostly present with adrenocortical lesions, whereas the cause of hypercortisolism in older children is usually a corticotropin (ACTH) secreting pituitary adenoma. Bilateral Nodular Adrenal Hyperplasia (BNAH) may be a component of a genetic condition such as Carney complex and isolated BANH is very rare. We present an uncommon case of ACTH-independent CS in an adolescent girl.
Clinical Presentation: The patient was admitted with weight gain. She had truncal obesity, moon facies and mild hypertrichosis, but no hypertension, no striae and no skin pigmentation. Her height Standard Deviation Score (SDS) was below -2. Laboratory investigation revealed elevated plasma and urinary cortisol and normal ACTH levels. Overnight 1 mg dexamethasone and lowdose dexamethasone tests failed to supress cortisol. Imaging studies revealed nodular hyperplasia of the left adrenal gland. The patient underwent bilateral laparoscopic adrenalectomy and histological evaluation revealed diffuse proliferation of right adrenal gland and micronodular proliferation in addition to diffuse proliferative areas in left adrenal gland. The surgery was curative and the patient was put on glucocorticoid and mineralocorticoid replacement therapy.
Keywords: Cushing syndrome; Bilateral adrenal hyperplasia; Adrenal nodules; Adolescent; Laparoscopic adrenalectomy
Abbreviations
ACTH: Corticotrophin; BNAH: Bilateral Nodular Adrenal Hyperplasia; CD: Cushing Disease; CS: Cushing Syndrome; CT: Computerized Tomography; MRI: Magnetic Resonance Imaging; PPNAD: Primary Pigmented Nodular Adrenocortical Disease; SDS: Standard Deviation Score; RR: Reference Range
Introduction
Cushing Syndrome (CS) is a rare clinical entity in children which is characterized by glucocorticoid excess. The most common cause of CS is exogenous administration of glucocorticoids or corticotropin (ACTH). On the other hand, the term “Cushing Disease (CD)” describes hypercortisolism due to an ACTH-secreting pituitary adenoma and is the most common cause of endogenous CS. CD comprise 75-80% of pediatric cases in children over 5 years of age [1]. In younger children, CS may be caused by ACTH-independent cortisol producing adrenocortical lesions including adrenal adenoma, adrenal carcinoma or adrenal hyperplasia (diffuse or multinodular). These conditions are reported to be the cause of CS as many as half of CS cases in children younger than 7 years of age [2]. Ectopic production of ACTH is particularly rare in paediatric age group.
ACTH-independent multinodular adrenal hyperplasias may be divided in two groups based on the size of the nodules, with macronodular disorders associated with nodules larger than 1 cm and micronodular hyperplasias associated with nodules that are smaller than 1 cm [2]. Macronodular adrenal hyperplasias are usually related to McCune-Albright syndrome (OMIM 174800) in children. This condition occurs due to somatic mutations in the GNAS1 gene. Benign micronodular hyperplasias include Primary Pigmented Nodular Adrenocortical Disease (PPNAD- OMIM 610489) and isolated micronodular adrenal disease. PPNAD usually accompanies to the “Carney complex” (OMIM 160980), which is a rare multiple neoplasia syndrome depicted by pigmented lentigens, myxomas, schwannomas and various endocrine tumors [3]. Inactivating mutations of the PRKAR1A gene have been described in the majority of cases with Carney complex. Recently, in patients with Cushing syndrome caused by bilateral adrenocortical lesions who have neither GNAS nor PRKAR1A gene mutations, inactivating mutations in phosphodiesterase gene family have been identified [4]. In this report we describe an adolescent girl with Cushing syndrome caused by isolated bilateral micronodular adrenal disease who has been cured by bilateral laparoscopic adrenalectomy. We want to emphasize the importance of clinical suspicion in the diagnosis of this rare condition in this age group.
Case Presentation
12 years and 7 months old female patient was admitted with excess gain of weight. She was born 3200 grams to nonconsangenious parents at term, her developmental milestones were normal. Her weight has been always normal but she gained more than 15 kilograms for the last year although she had no significant changes in her eating habits and daily exercise. On physical examination, her systemic arterial blood pressure was 90/60 mmHg, height: 138.5 cm (-2.91 SDS), weight: 47 kg (-0.15 SDS), body mass index: 24.5 kg/m² (+1.49 SDS). She had truncal obesity, moon facies and mild hypertrichosis. Her pubic hair and breast development were stage 4 and stage 3 respectively, according to Tanner staging. No stretch marks were noticed. Physical examination of all other systems was normal. Laboratory measurements revealed mildly elevated morning cortisol levels (25.92 μg/dL, Reference range [RR]:3-10) with normal fasting glucose (71 mg/dl, RR:74-110) and insulin (13.53 μU/mL, RR: 0-17) and elevated serum lipids (Total Cholesterol: 267 mg/dL, RR: 0-200, Low Density Lipoprotein-Cholesterol: 167 mg/dL, RR: 0-130, High Density Lipoprotein: 66 mg/dL, RR: >40 and Triglyceride: 169 mg/dL, RR: 0-150). Other laboratory findings were within normal limits. The patient was further evaluated for hypercortisolism and hormonal investigation revealed loss of circadian rhythm (plasma morning cortisol 42.17 μg/dL (RR:3-10), midnight cortisol 16.44 μg/ dL (RR: <7.5) and morning ACTH 6.74 pg/ml (RR: 2-49), midnight ACTH: 7.97 pg/ml (RR 5-10, normally lower than morning level) and elevated Urinary Free Cortisol (UFC) level (573.3 mg/day [398 μg/ m²/day], RR: 10-100 μg/day). Overnight Dexamethasone (DXM) test showed no suppression (plasma cortisol after 1 mg DXM 24.96 μg / dL, RR: <1.8). Low dose DXM suppression test (0.5 mg every 6 hours for 8 doses) showed no suppression of cortisol levels (plasma cortisol 10.52 μg/dL, RR: <1.8, UFC 110 μg /day [76.38 μg /m²/day], normally < 10% of baseline, ACTH: <1.0 pg/ml after test).
The Computerized Tomography (CT) of the adrenals revealed diffuse thickening of the medial cruris of the left adrenal gland, whereas the right adrenal gland appeared normal in size. A Magnetic Resonance Imaging (MRI) with suppression of fat signal technique was obtained and it revealed nodular hyperplasia of the left adrenal gland with normal manifested right adrenal gland.
The hormonal and radiological findings confirmed the diagnosis of ACTH-independent CS and we decided to perform the adrenalectomy with laparoscopic approach. On surgery, both adrenals were enlarged. Macroscopically right adrenal was measured 15 grams and 5x4x3 cm, left adrenal was measured 23 grams and 7x3x2.5 cm. In cut section, both right and left adrenal gland revealed diffuse hyperplasia in focal areas of adrenal glands (Figure 1). There were neither macronodules nor pigmented areas. Histological assessment revealed diffuse proliferation of cells with uniform small round nuclei and clear cytoplasm cell showing alveolar and trabecular pattern in right adrenal gland (Figure 2A). In left adrenal gland, micronodular proliferation in addition to diffuse proliferative areas was detected (Figure 2B).

Figure 1: Diffuse hyperplasia of the right and left adrenal glands (macroscopic
view).
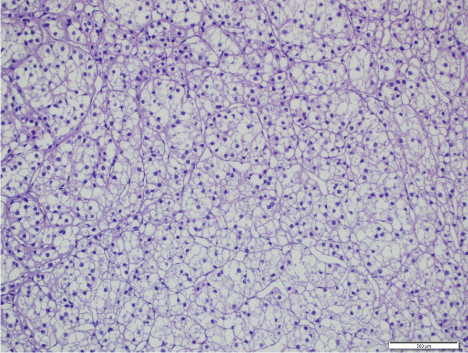
Figure 2A: Diffuse proliferation of cells with alveolar and trabecular pattern in
the right adrenal gland (H&Ex100).
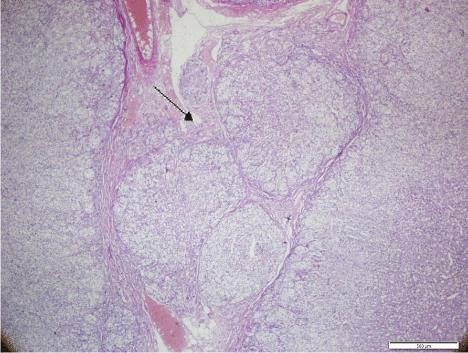
Figure 2B: Micronodular proliferation in addition to diffuse proliferative areas
in the left adrenal gland (arrow) (H&Ex20).
The post-operative course of our patient was uneventful. She received glucocorticoid and mineralocorticoid replacement therapy during and after surgery. On six months follow-up the patient showed improvement in the laboratory and clinical findings of CS.
Discussion
We have described a rare case of ACTH-independent CS. The most common form of endogenous CS in adolescents is ACTHsecreting pituitary adenoma. Yet our patient had ACTH-independent CS which is less frequent and mostly seen in infants and young toddlers. Moreover, autonomous secretion of cortisol from adrenals is usually due to isolated, solitary adenoma and bilateral diseases are rarely seen in paediatric age. In such cases, a good family history should be taken and systemic investigation should be made in order to exclude genetic conditions associated with bilateral adrenal hyperplasia like Carney complex and McCune Albright syndrome. Our patient had neither family history nor skin pigmentation. She underwent cranial MRI scan and echocardiography and both were ordinary. Her adrenal glands were not pigmented consequently our patient was diagnosed with isolated micronodular disease. A recent review suggests that bilateral nodular adrenal disease in children is more common than previously thought [2].
Our patient had some of the clinical findings of CS like weight gain, moon facies and hypertrichosis. But other symptoms and signs of CS such as skin atrophy, violaceous striae, buffalo hump, hypertension and muscle weakness were lacking. The onset of the disease was insidious in our patient and initial serum cortisol level was not very elevated and ACTH level was not supressed. We could overlook CS in this patient because her clinical and laboratory findings could easily be implicated as simple obesity. The short stature with obesity led us to the suspicion of CS in our patient. It is reported that lack of height gain consistent with the weight gain is the most common presentation of CS in children [5]. The paediatricians should not solely rely on the clinical and laboratory findings in obese patients with short stature and CS should be kept in mind.
The imaging studies may be useful in the diagnosis of ACTHindependent CS. Adrenal CT with contrast has been identified as the most diagnostic imaging tool for cortical lesions therefore CT should be preferred over MRI [5]. In our patient the CT scan showed hyperplasia in left adrenal gland and the right gland was normal. We performed a fat-supressed MRI which offered us a better vision of the nodular pattern of the left adrenal gland although the right adrenal gland was normal. Fat suppressed MRI is used in order to eliminate artifacts and has been reported as advantageous in imaging of the adrenal gland [6]. Yet, both CT and MRI failed to demonstrate the hyperplasia of the right adrenal gland in our patient. The determination of the diagnosis is crucial before surgery to avoid re-operation after unilateral adrenalectomy.
One might question why we have decided to remove both adrenals although the right adrenal appeared normal on imaging studies. Powell, et al. [7] reviewed their experience in the surgical management of 34 patients with micronodular hyperplasia. They concluded that laparoscopic bilateral resection was curative in their patients. In that study three patients underwent unilateral resection and neither patient was cured biochemically. Zografos, et al. [8] reported a case with PPNAD who underwent unilateral adrenal resection but further needed another surgery six months later for the other adrenal due to recurrence of the disease. In our patient the right gland was observed hyperplasic during surgery which encouraged us to decide bilateral resection. In fact, the histologic examination confirmed the diagnosis of bilateral disease. Laparoscopic adrenalectomy is associated with lower morbidity rate compared with open resection and it also provides shorter hospitalization and less pain after surgery [7,8].
In conclusion, CS should be considered in patients with obesity accompanied with short stature even though the clinical picture is subtle. Bilateral laparoscopic adrenalectomy is the preferred standard of care for CS due to micronodular hyperplasia.
Conclusion
Although rare, BNAH should be considered in adolescent patients with obesity accompanied with short stature and bilateral laparoscopic adrenalectomy is the preferred treatment.
References
- Savage MO, Storr HL. Pediatric Cushing’s disease: Management Issues. Indian J Endocrinol Metab. 2012; 16: 171-175.
- Stratakis CA. Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin-independent Cushing syndrome). Endocr Dev. 2008; 13: 117-132.
- Almeida MQ, Stratakis CA. Carney complex and other conditions associated with micronodular adrenal hyperplasias. Best Pract Res Clin Endocrinol Metab. 2010; 24: 907-914.
- Carney JA, Gaillard RC, Bertherat J, Stratakis CA. Familial micronodular adrenocortical disease, Cushing syndrome and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A). Am J Surg Pathol. 2010; 34: 547-555.
- Stratakis CA. Cushing Syndrome in Pediatrics. Endocrinol Metab Clin North Am. 2012; 41: 793-803.
- Boland GW, Lee MJ. Magnetic resonance imaging of the adrenal gland. Crit Rev Diagn Imaging. 1995; 36: 115-174.
- Powell AC, Stratakis CA, Patronas NJ, Steinberg SM, Batista D, Alexander HR, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery. 2008; 143: 750-758.
- Zografos GN, Pappa T, Avlonitis S, Markou A, Chrysikos DT, Kaltsas G, et al. Primary pigmented nodular adrenocortical disease presenting with a unilateral adrenocortical nodule treated with bilateral laparoscopic adrenalectomy: a case report. J Med Case Rep. 2010; 4: 230.