
Case Report
J Pediatri Endocrinol. 2017; 2(1): 1014.
Hypopituitarism may be an Additional Feature of SIM1 and POU3F2 Containing 6q16 Deletions in Children with Early Onset Obesity
Rutteman B¹, De Rademaeker M², Gies I¹, Van den Bogaert A², Zeevaert R¹, Vanbesien J¹ and De Schepper J¹*
¹Department of Pediatric Endocrinology, UZ Brussel, Belgium
²Department of Genetics, UZ Brussel, Belgium
*Corresponding author: De Schepper J, Department of Pediatric Endocrinology, UZ Brussel, Belgium
Received: December 23, 2016; Accepted: February 21, 2017; Published: February 22, 2017
Abstract
Over the past decades, 6q16 deletions have become recognized as a frequent cause of the Prader-Willi-like syndrome. Involvement of SIM1 in the deletion has been linked to the development of obesity. Although SIM1 together with POU3F2, which is located close to the SIM1 locus, are involved in pituitary development and function, pituitary dysfunction has not been reported frequently in cases of 6q16.1q16.3 deletion involving both genes. Here we report on a case of a girl with typical Prader-Willi-like symptoms including early-onset hyperphagic obesity, hypotonia, short hands and feet and neuro-psychomotor development delay. Furthermore, she suffered from central diabetes insipidus, central hypothyroidism and hypocortisolism. Her genetic defect is a 6q16.1q16.3 deletion, including SIM1 and POU3F2. We recommend searching for a 6q16 deletion in children with early onset hyperphagic obesity associated with biological signs of hypopituitarism and/or polyuria-polydipsia syndrome. The finding of a combined SIM1 and POU3F2 deletion should prompt monitoring for the development of hypopituitarism, if not already present at diagnosis.
Keywords: 6q16 deletion; SIM1; POU3F2; Prader-Willi-like syndrome; Hypopituitarism
Introduction
Prader-Willi-Syndrome (PWS), one of the most common genetic causes of early onset obesity, is characterized by neonatal hypotonia and feeding problems, as well as delayed neuro-psychomotor development and early childhood-onset hyperphagic obesity. In the absence of chromosome 15 abnormalities, this phenotype is called Prader-Willi-Like Syndrome (PWLS) and has been observed in deletions of chromosomes 1p, 2p, 3p, 6q, 9q, 10q and 12q and in different X-chromosome abnormalities, such as inversion and Xq deletions [1,2].
Frequently described chromosomal abnormalities in PWLS are 6q16 deletions. Haploinsufficiency of the Single-minded homolog 1 (SIM1) gene and the POU Domain, class 3, transcription factor 2 (POU3F2), located in respectively the 6q16.3 and 6q16.1 region, have been suggested to play a role in early-onset hyperphagic obesity both through hyperphagia and reduced energy expenditure [3-5]. POU3F2 and SIM1 act in a cascade for Arginine-Vasopressin (AVP), Thyrotropin-Releasing Hormone (TRH) and Corticotropin- Releasing Hormone (CRH) neuron differentiation in the supraoptic and paraventricular nuclei [6]. Sim1 is a basic helix-loop-helix PAS transcription factor, involved in the development of the neurons of the supraoptic, paraventricular and anterior periventricular nuclei of the rodent hypothalamus, which produce the neuro-endocrine peptides oxytocin, AVP, CRH, TRH and somatostatin [6,7]. Pou3f2 is highly expressed in the hypothalamus and is involved in the vasopressin, oxytocin and gonadadotropin releasing hormone expression in the rodent hypothalamus [8]. Based on their role in hypothalamic development, one would expect the occurrence of central diabetes insipidus and of some specific pituitary hormone deficiencies in children with a combined SIM1 and POU3F2 deficiency. We describe the clinical and hormonal findings in a 10 year old female child with a relatively small deletion of 6q16.1q16.3, containing the SIM1 and POU3F2 loci, who presented with early-onset hyperphagic obesity, central diabetes insipidus and secondary hypothyroidism and hypocortisolism.
Case Report
A 10 year old girl with early-onset hyperphagic obesity and mental retardation was seen at our endocrine clinic for additional endocrine investigations because of unexplained hypothyroxinemia and hypocortisolism.
Her parents were not consanguineous. Her father was known with persistent enuresis nocturna and his Body Mass Index (BMI) was 35.9 kg/m2. Her mother had a gastric bypass at the age of 36 years, decreasing her BMI from 53.9 kg/m2 to 25 kg/m2. Her only brother and paternal grandparents had severe overweight as well.
She was born at 40 weeks of gestation with a normal birth weight (3.520 kg) and birth length (50.5 cm). Her psychomotor development was slightly delayed (sitting at 8 months and first words at 16 months). She presented with increasing behavioral problems since infancy (mood swings, intolerance to frustration, aggressiveness, emotional liability). Because of learning difficulties, she received special education (type 8 in the Flemish school system). She presented with hyperphagia and increasing body weight from the first year of life (Figure 1). From the age of four years, she had polydipsia (drinking more than 4 liters a day), polyuria and persisting enuresis nocturna.
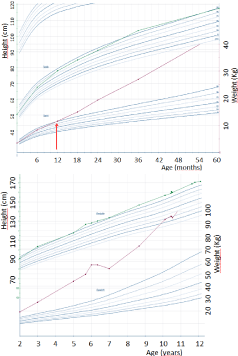
Figure 1: Growth curves showing onset (arrow) of obesity at age 12 months
and continuing through childhood. Weight in red and length in green.
Previous hormonal and metabolic investigations between the age of 5 and 9 years showed a partial urinary concentration deficit during a water deprivation test (max urinary osmolality: 529mosm/kg H2O), a low FT4 concentration of 9.3 pmol/L (reference range 12-22) with a normal TSH concentration, and insulin resistance on oral glucose tolerance testing (peak insulin 210 mIU/L). Previous genetic studies included a negative genetic screening for Prader-Willi Syndrome (PWS) and MC4R gene mutations. Brain MRI imaging was normal. Dietary counseling was given, but her overweight increased further. During follow-up, low FT4 (<10 ng/l) and cortisol concentrations (<60 μg/L) were found.
At the age of 10 years, physical presentation showed body weight of 96 kg (+7SD), height of 161 cm (+3SD), head circumference of 56 cm, BMI of 38.0 kg/m2, blood pressure of 147/96 mmHg, pulse of 75 per minute, and Tanner stage A1P1M1. Vulva and clitoris were normal. Facial dysmorphic features including a flat midface, bilateral epicanthal folds and deep set eyes were noticed. She had hyperlordosis, bilateral genu valgus, short hands with tapering fingers, short feet and global hypotonia (Figure 2).
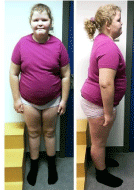
Figure 2: Photographs of the proband showing obesity, facial dysmorphic
features (a flat midface, bilateral epicanthal folds and deep set eyes),
hyperlordosis, bilateral genu valgus, short hands with tapering fingers and
short feet.
Bone age was 13 year (Greulich and Pyle method). Biological and hormonal investigations showed an increased uric acid (8.3 mg/L) and confirmed the low FT4 (9.4 pmol/L) and a low morning cortisol (47 μg/L), with a non-compensatory low- normal ACTH (13.8 ng/L) (Table 1). A TRH test showed a high TSH reserve (peak TSH 39.3 mIU/L with prolonged response). A CRH test showed an exaggerated ACTH response (65.8 ng/L). She was treated with oral levothyroxine 100 μg once daily and intranasal desmopressin 10 μg twice daily.
![]()
Analyte
Value
Reference range
TSH (mIU/L)
4.34
0.27-4.2
Free T4 (pmol/L)
9.4
12.0-22.0
LH (IU/L)
<0.1
0.10-11.9
FSH (IU/L)
0.9
2.11-11.1
Prolactin (mcg/L)
3.72
1.97-31.44
ADH (ng/L)
<0.4
<2.0
IGF-1 (mcg/L)
78
75-440
IGFBP-3 (mcg/L)
4208
2321-6195
ACTH (ng/L)
13.8
Jun-55
8 am Cortisol (mcg/L)
47.1
62-194
CBG (mg/L)
58.4
31.0-53.4
17-OH progesteron (mcg/L)
<0.1
0.21-1.4
DHEAS (mg/L)
0.3
0.34-2.80
Estradiol (ng/L)
<5
<40.5
Androstenedion (ng/L)
542
<2540
SHBG (mmol/L)
15.1
20.0-99.6
Table 1: Hormonal results at the age of 10 years. TSH: Thyroid Stimulating Hormone, LH: Luteinizing Hormone, FSH: Follicle Stimulating Hormone, ADH: Anti-Diuretic Hormone, IGF-1: Insulin-Like Growth Factor 1, IGFBP-3: Insulin- Like Growth Factor Binding Protein 3, ACTH: Adrenocorticotropic Hormone, CBG: Cortisol Binding Globulin, DHEAS: Dehydroepiandrosteron Sulfate, SHBG: Sex Hormone Binding Globulin.
An array comparative genomic hybridization revealed a 6.9 Mb deletion on chromosome 6q (arr [hg 19] 6q16.1q16.3 (96,037,310- 102,931,814)x1 dn). The genes with OMIM reference involved in the deletion include: MANEA, FUT9, UFL1, FHL5, GPR63, NDUFAF4, KLHL32, MMS22L, POU3F2, FBXL4, FAXC, COQ3, PNISR, USP45, CCNC, PRDM13, MCHR2, SIM1, ASCC3 and GRIK2 [9]. Genetic investigation of the parents showed a normal array CGH.
Discussion
A relatively small deletion of 6q16.1q16.3, involving both SIM1 and POU3F2, was documented in a severely obese girl, who presented with the typical clinical features of PWLS, including hyperphagic early-onset obesity, hypotonia, short hands and feet and neuropsychomotor development delay. Furthermore, she had several features that have been described previously in cases with 6q16 deletions, such as behavioral problems and non-PWS associated facial dysmorphic features, including a flat midface, bilateral epicanthal folds and deep set eyes. In addition, central diabetes insipidus, central hypothyroidism and hypocortisolism were observed.
In similar and other 6q deletions of variable sizes, whereas obesity has been a common finding, hormonal deficiencies have been rarely reported. Hypothyroidism of unclear origin has been described in five cases [10-12], evolving hypopituitarism, but of childhood onset, was documented in one case [13] and GH deficiency in another child [10]. In 2000, Holder and colleagues reported hypocortisolism in a patient with severe early onset obesity and a de novo balanced translocation between chromosomes 1p22.1 and 6q16.2 [3].
In most cases with a deletion of only the 6q16.1q16.3 region, as in our case, hyperphagia and obesity started in general at young age. In cases with only POU3F2 deletions, hyperphagia was reported to develop after the onset of obesity [5]. Both central hypothyroidism and hypocortisolism was documented in our case. Growth hormone testing was not performed given the regular linear growth above percentile 97 and the normal serum IGF-1 concentration. In addition, Sim1-/-or Pou3f2-/-mouse do not have GHRH deficiency. Given the age of the patient, hypogonadotropic hypogonadism could not be assessed. Of the 12 cases with a 6q16.1q16.3 deletion including both SIM1 and POU3F2 previously reported in the literature [10- 12,14,15], only one case had symptoms of hypogonadism [11] and another case had enuresis nocturna [10], but none had been tested for either gonadotropin or ADH deficiency. None of the above 12 mentioned cases had a formal anterior or posterior pituitary function evaluation (Table 2, with arrayCGH results in supplementary Table S1). We suspect that behavioral problems of these children might have suspended detailed hormonal testing. In addition, increased drinking might have been attributed to their associated aberrant behavior. Furthermore, the pituitary hormones might have only been partially deficient, as was documented in our patient. We wanted to exclude AVP, TRH and CRH deficiency in our patient since, at least in mice, Pou3f2 and Sim1 act in a cascade for AVP, TRH and CRH neuron differentiation in the supraoptic and paraventricular nuclei [6,7]. In Sim1 heterozygous mice, hypothalamic mRNA levels of TRH, AVP and CRH were 42, 41 and 30% respectively [16] and in Pou3f2 heterozygous mice decreased AVP expression was evidenced [8]. Thus the combination of POU3F2 and SIM1 deficiency might explain the central diabetes insipidus, central hypothyroidism and central hypocortisolism in our patient. Based on our findings, it seems that hypopituitarism may be a feature of children with small 6q16 deletions, which might have been underestimated for several reasons up to now.
![]()
Present case
Rosenfeld
Bonaglia
El Khattabi (case 2)
El Khattabi (case 3)
El Khattabi (case 4)
El Khattabi (case 7
El Khattabi (case 10)
El Khattabi (case 11)
El Khattabi (case 12
El Khattabi (case 13)
Wang
Le Caignec
Total
Total %
Sex
F
F
M
F
M
M
M
M
M
M
M
F
F
-
-
Age last evaluation
10y
3y
16y
17y
3,5y
23y
18y
8y
12y
10y
7y
7y
2y
-
-
Hypotonia
+
-
+
-
+
?
-
-
-
?
-
-
+
4/10
40%
Feeding problems
-
?
+
-
-
?
-
-
-
+
+
-
?
3/9
33%
Hyperphagia
+
?
?
+
+
?
?
+
?
?
-
-
-
4/7
57%
Obesity
+
+
overweight
+
+
+
+
+
overweight
overweight
overweight
+
-
8/13
62%
Onset
1y
?
?
?
?
?
?
?
?
?
?
?
-
-
-
Motor delay
+
+
+
+
+
?
+
+
+
+
+
+
+
12/12
100%
Mental retardation
+
+
+
+
+
+
+
+
+
+
+
+
+
13/13
100%
Behavioural problems
+
-
+
+
+
?
+
-
-
?
+
-
+
7/11
64%
Facial dysmorphisms
+
-
+
+
+
+
+
+
+
+
+
+
+
12/13
92%
Round face
+
-
+
+
+
+
+
+
+
?
?
-
+
9/11
82%
Macrocephaly
-
+
-
+
+
-
-
-
-
+
+
+
-
6/13
46%
Bulbous nose
-
-
-
+
+
-
-
-
+
-
+
+
+
6/13
46%
Philtrum
-
-
short
marked
-
-
-
-
marked
-
long
-
prominent
-
-
Other
-
-
-
horizontal eyebrows
-
-
-
synophris
epicanthal folds
-
hypertelorism
synophris
hypertelorism
-
-
Acromicria
+
-
-
+
-
-
-
+
+
-
-
-
?
5/12
42%
Brain MRI
normal
small pituitary gland
enlarged third ventricle and
hypertrophy of the left choroid plexus?
?
?
?
normal
normal
ventriculo- megaly
normal
?
normal
-
-
Visus problems
-
-
?
severe myopia
-
-
myopia
astigmatism
hypermetropia
nystagmus
-
-
strabismus, hypermetropia
-
-
Pituitary dysfunction
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
Thyrotropin deficiency
+
+
?
?
?
?
?
?
?
?
?
?
?
-
-
Corticotropin deficiency
+
?
?
?
?
?
?
?
?
?
?
?
?
-
-
Central diabetes insipidus
+
?
? (enuresis +)
?
?
?
?
?
?
?
?
?
?
-
-
GH deficiency
-
?
?
?
?
?
?
?
?
?
?
?
?
-
-
Gonadotropin deficiency
prepubertal
?
?
?
?
?
?
?
hypogonadism
?
?
?
?
-
-
Table 2: Clinical features and additional results of reported cases with a similar deletion (deletion of only 6q16.1q16.3 and including SIM1 and POU3F2).
Based on this case report and review of literature, we recommend searching for a 6q16 deletion in children with early onset hyperphagic obesity associated with biological signs of hypopituitarism and/ or polyuria-polydipsia syndrome. We suggest the use of molecular karyotyping with overlapping deletions to delineate critical regions, thereby improving genotype/phenotype correlation. On the other hand, the finding of a combined SIM1 and POU3F2 deletion should prompt monitoring for the development of hypopituitarism, if not already present at diagnosis.
References
- Varela MC, Simoes-Sato AY, Kim CA, Bertola DR, De Castro CI, Koiffmann CP. A new case of interstitial 6q16.2 deletion in a patient with Prader-Willi-like phenotype and investigation of SIM1 gene deletion in 87 patients with syndromic obesity. Eur J Med Genet. 2006; 49: 298-305.
- Rocha CF, Paiva CL. Prader-Willi-like phenotypes: a systematic review of their chromosomal abnormalities. Genet Mol Res. 2014; 13: 2290-2298.
- Holder JL Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000; 9: 101-108.
- Xi D, Gandhi N, Lai M, Kublaoui BM. Ablation of Sim1 neurons causes obesity through hyperphagia and reduced energy expenditure. PLoS One. 2012; 7.
- Kasher PR, Schertz KE, Thomas M, Jackson A, Annunziata S, Ballesta-Martinez MJ, et al. Small 6q16.1 Deletions Encompassing POU3F2 Cause Susceptibility to Obesity and Variable Developmental Delay with Intellectual Disability. Am J Hum Genet. 2016; 98: 363-372.
- Michaud JL, Rosenquist T, May NR, Fan CM. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998; 12: 3264-3275.
- McCabe M, Dattani M. Genetic aspects of hypothalamic and pituitary gland development. Handb Clin Neurol. 2014; 124: 3-15.
- Shonemann M, Ryan A, McAVilly R, O'Connell S, Arias C, Kalla K, et al. Development and survival of the endocrine hypothalamus and the posterior pituitary gland requires the POU domain factor Brn-2. Genes Dev. 1995; 9: 3122-3135.
- Online Mendelian Inheritance in Man (OMIM).
- Bonaglia MC, Ciccone R, Gimelli G, Gimelli S, Marelli S, Verheij J, et al. Detailed phenotype-genotype study in five patients with chromosome 6q16 deletion: narrowing the critical region for Prader–Willi-like phenotype. Eur J Hum Genet. 2008; 16: 1443-1449.
- El Khattabi L, Guimiot F, Pipiras E, Andrieux J, Baumann C, Bouquillon S, et al. Incomplete penetrance and phenotypic variability of 6q16 deletions including SIM1. Eur J Hum Genet. 2015; 23: 1010-1018.
- Rosenfeld JA, Amrom D, Andermann E, Andermann F, Veilleux M, Curry C, et al. Genotype-phenotype correlation in interstitial 6q deletions: a report of 12 new cases. Neurogenetics. 2012; 13: 31-47.
- Izumi K, Housam R, Kapadia C, Stallings VA, Medne L, Shaikh TH, et al. Endocrine phenotype of 6q16.1-q21 deletion involving SIM1 and Prader-Willi syndrome-like features. Am J Med Genet A. 2013; 161A: 3137-3143.
- Le Caignec C, Swillen A, Van Asche E, Fryns JP, Vermeesch JR. Interstitial 6q deletion: clinical and array CGH characterization of a new patient. Eur J Med Genet. 2005; 48: 339-345.
- Wang J, Turner L, Lomax B, Eydoux P. A 5-Mb micro deletion at 6q16.1q16.3 with SIM1 gene deletion and obesity. Am J Med Genet A. 2008; 146A: 2975-2978.
- Tolson KP, Gemelli T, Meyer D, Yazdani U, Kozlitina J, Zinn AR. Inducible Neuronal Inactivation of Sim1 in Adult Mice Causes Hyperphagic Obesity. Endocrinology. 2014; 155: 2436-2444.